R语言学习-富集分析泡泡图刚一出品,Y叔就说有硬伤。Y叔是著名富集分析软件clusterprofiler的原创,而且软件内集成dotplot,enrichmap,cnetmap (后续也实现这两个的一步出图)等画图方法,具体看这个教程http://guangchuangyu.github.io/2016/01/go-analysis-using-clusterprofiler/或Biobabble公众号。
这个意见得重视,不过大夏天的,锅还是不能背着,回应下Y叔的回应。
一个command出图,小白上点心可以的
图是ggplot2画的(散点图),在脚本功能描述里有写。只需要在终端输入脚本的名字sp_enrichmentPlot.sh
(脚本需在环境变量中)就可以看到脚本功能描述、输入文件样例、参数列表和使用例子。
目前脚本的输入是不支持Windows的Excel格式的,只支持TAB分割的文本文件。之前一直在Linux下工作,文件名都不写后缀的。后来跟Windows用户打交道多,为了方便他们打开,强行加了xls后缀,这一点有误导。
脚本依赖R,这是应该做一个判断和提示的。但有R却提示ggplot2包或其它包不存在,是可以用-i TRUE来自动安装包的。(最近在跟德春合作,完善包的自动检测和安装,最后一起整合在分发包。)
Y叔的这两点确实戳中了当前文档信息不完善的弱点,也希望大家多反映使用过程中的问题,帮助我们改进文档。
不过话说回来,如果仔细看了当前脚本的功能描述和参数提示,上点心得小白不只可以一步画图,还可以随意调整样式。
不只clusterprofiler的用户需要
如果用clusterprofiler做富集分析,write.table输出结果,那么输入文件、R、ggplot2都有了,一步出图没有问题。(Windows下没bash?https://mp.weixin.qq.com/s/g1twNEsPWZb_tZFDyoTqVQ,看这里)
Y叔说既然有了clusterprofiler,一步步在R里面做不是很好,为啥要把数据导出来?
首先,转换数据、存储数据这一步是独立于作图的。图只能显示部分数据(这点Y叔也有提到,用了simplify会好些,但也还会有不少通路),所有富集条目导出作为文章附表,以显示信息的全面和真实。
其次,即便可以画出所有富集数据(用一副大图),也会先对结果做下筛选,一些特别基础的、极父层的生物富集通路也会选择不展示,优先展示样品属性更相关的。毕竟是看在哪些通路里面富集,不是看在哪些通路里面最富集。所以需要导出数据,做下筛选,然后一步绘图。
再次,不少人拿到的是已经做好的富集分析结果(可能是clusterprofiler的,也可能是其它软件的),总不能再导入clusterprofiler绘图吧,可以有更简单的方式的。
最后,一个图做出来,需要反复修改。今天用clusterprofiler做了富集分析,运用dotplot配合不同参数出了图;过几天心情一变,想换个风格了,怎么办?再运行一次clusterprofiler还是加载之前存储的.Rdata。好像都不太方便,还是用导出的文本一步出图吧。
关于硬伤
示例图中没有overlap不是绘图脚本的问题,是数据筛选的问题。脚本如实反映筛选出的数据。如果选择的通路在一个基因集合富集,另外一个不富集,就不显示;如果选择的通路是在多个基因集中都富集的,但富集程度不同,是可以显示的;而且这些通路会优先显示在图的上部,下面我会给出例子。
一步出图也可以定制
说到互动,一步出图不只可以,而且还可以记录互动(给每个输出文件加个唯一的字符串做为文件名的一部分就可以了,不过这个没用到,之前就没写这个参数)。
拿Y叔的数据做个例子
library(clusterProfiler)
library(dplyr)
data(gcSample)
x <- compareCluster(gcSample,'enrichDO')
#获取数据,取出每个组最富集的10个条目,存储起来
#不麻烦的吧
x <- x@compareClusterResult
y = x %>% group_by(Cluster) %>%top_n(-10, pvalue)
y = x[x$Description %in% y$Description,]
在样品名中包含差异基因的数目
#这个不解释了,前面绘图教程中提到过
y$Sample <- paste0(y$Cluster,"(",unlist(lapply(strsplit(y$GeneRatio, '/'), function(x)x[2])),")")
#这次存成tsv,省得误会了
write.table(y,file="enrichment.tsv", sep="\t", col.names=T, row.names=F,quote=F)
首先出第一个图,很简单,一个命令,设置下颜色。
+更正下,我这里作弊了一下,之前点的大小都是Gene Count这样的纯数字列来显示的,不支持GeneRatio这样的分数形式的列(上一版本,如果GeneRatio出现在横轴是支持的),做了下修改,也支持了。如果想用之前的代码重复,把-s GeneRatio改为-s Count。
#具体参数解释看上一篇公众号文章
# -C:指定颜色,默认是红绿,可以是任何两个R支持的颜色名字
sp_enrichmentPlot.sh -f enrichment.tsv -oSample -T string -v Description -c p.adjust -s GeneRatio -C"'red','blue'" -w 16
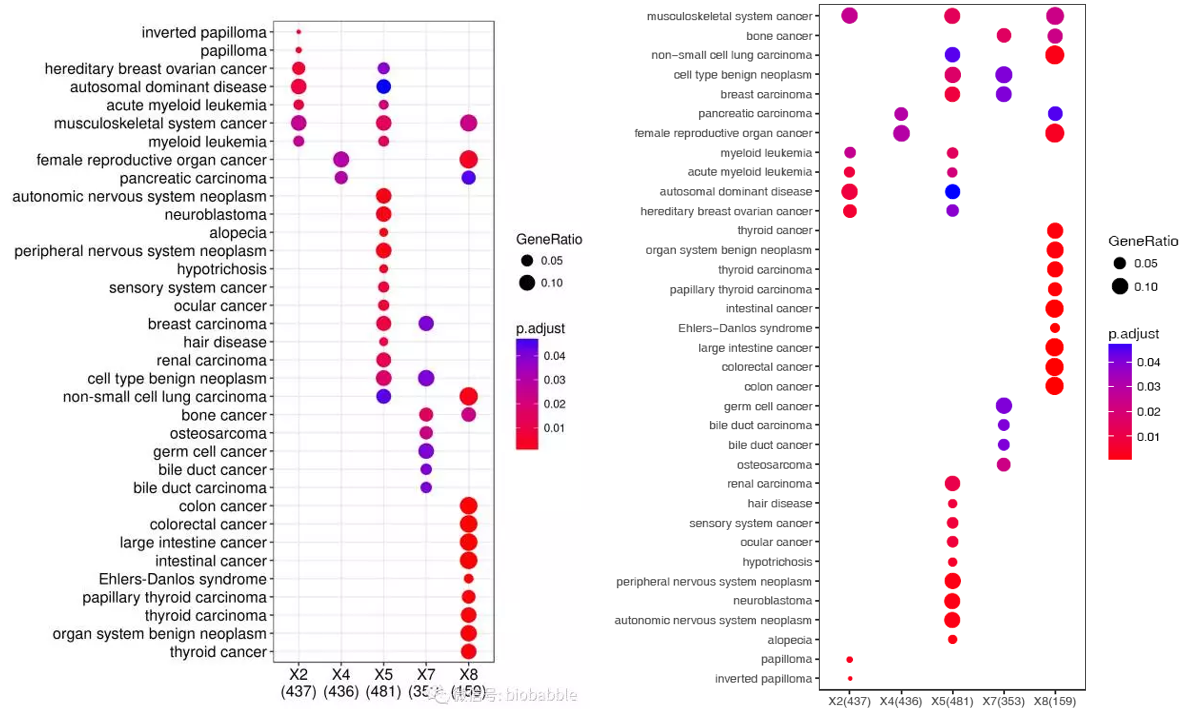
图略有不同,是因为排序的方式不同。sp_enrichmentPlot.sh设置的是先按照出现在不同样品最多的条目优先的策略,可以清晰的看到哪些是不同基因集共同富集的,哪些是不同基因集特异富集的。其它的都一样。当然没有设置字体参数,字体略小了些。
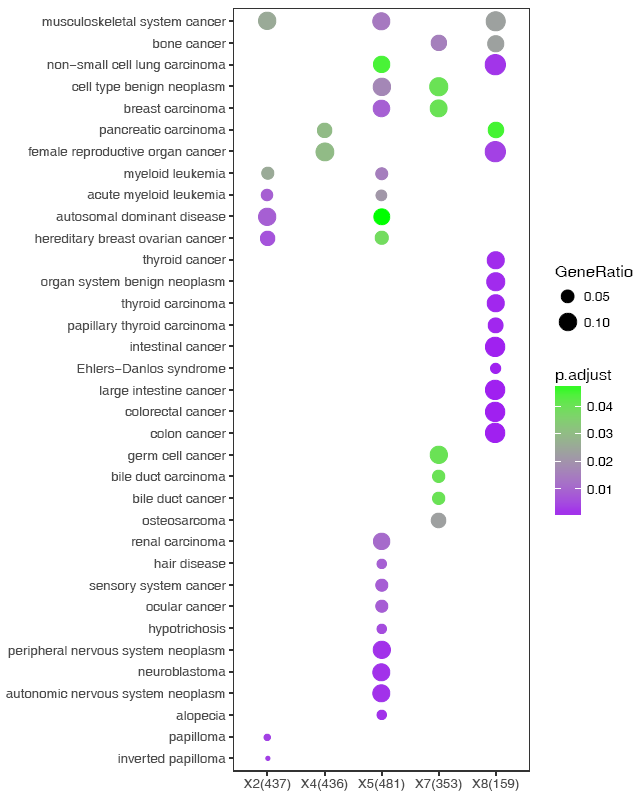
说还想要个紫绿的,也可以,只需改两个单词,而不是加一段代码scale_color_continuous(low='purple',
high='green')。
sp_enrichmentPlot.sh -f enrichment.tsv -oSample -T string -v Description -c p.adjust -s GeneRatio -C"'purple','green'" -w 16
改变下形状,也可以啊。程序中都留有一个参数可以写入ggplot2语句,就是下面的-z。那么,ggplot2能修改的样式也都可以。常用的修改会做成一个参数,不常用的就只能直接写命令了。能在Rstudio里面敲,那么也能在命令行敲。
#-z:后跟任何合法的ggplot2语句
sp_enrichmentPlot.sh -f enrichment.tsv -oSample -T string -v Description -c p.adjust -s GeneRatio -C"'purple','green'" -w 16 -z "+aes(shape=GeneRatio>0.1)"
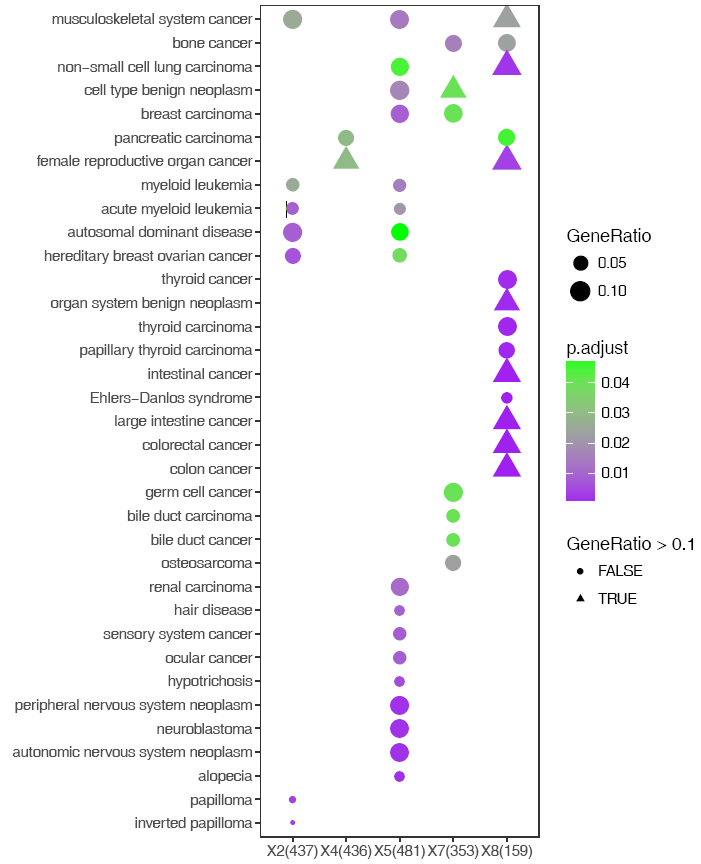
-z “+ any legal ggplot2command”
当然这一步也可以不通过增加ggplot2语句来实现,在数据中加一列就好了,通过-S指定参数。
不加ggplot2语句,只修改命令行参数,sp_enrichmentPlot.sh也可以调整图例的位置、输出文件的格式、是否对p.adj取log、是否分面等。
一步作图的优势
一步作图相比于直接写R代码有什么好处呢?
1.模块化好。也就是Y叔提到的数据处理和可视化分开;一步作图,只是作图,不做无关的处理,更随意。
2.易用性强。敲代码,总不如给改数来得快。
3.重用性强。假如我要做十几个分开的基因集合呢?一段段复制代码?改错了或忘记改某个地方了怎么办?
4.快速出图。先快速出个原型,再接着调整。













网友评论