常用的工具
PCA分析中常用的工具有GCTA中的PCA模块,老牌的软件EIGENSOFT中的smartpca,还有很多最近推出的R包都能够做PCA分析。然后分析完后,可视化的操作一般是使用R中ggplot等包去实现。
简单介绍一下GCTA和EIGENSOFT这两个工具:
GCTA
在群体遗传中,GCTA中的PCA模块是一款比较好的软件,不单可以做PCA的分析,其他LD,FST等一概都可以使用。工具官网http://cnsgenomics.com/software/gcta/#Overview 。这个软件能支持不同的平台,Windows,mac还有linux都有。
EIGENSOFT
这个工具是很经典老牌的工具,引用率已经过6000了,是非常可靠也得到了学术界认可的一款软件。工具官网:https://www.hsph.harvard.edu/alkes-price/software/, 也可以通过conda下载比较简便。这个工具的缺点就是它只支持linux系统,而且对输入文件的格式有一定的要求。
实战
实战一:GCTA-PCA
首先使用plink将vcf 格式的文件转化成所以.bed,.bim,.pam结尾的输入文件。
~/biosoft/plink --allow-extra-chr --out myplink_test --recode --vcf Gpan.prune.in.vcf
~/biosoft/plink --allow-extra-chr --file myplink_test --noweb --make-bed --out tmp
利用GCTA进行PCA构建
~/biosoft/gcta_1.91.6beta/gcta64 --bfile tmp --make-grm --autosome --out tmp
~/biosoft/gcta_1.91.6beta/gcta64 --grm tmp --pca --out pcatmp
运行玩后会生成三个文件
-rw-r----- 1 hhu pawsey0149 1.1K Oct 5 12:32 pcatmp.log
-rw-r----- 1 hhu pawsey0149 8.0K Oct 5 12:32 pcatmp.eigenval
-rw-r----- 1 hhu pawsey0149 45K Oct 5 12:34 pcatmp.eigenvec
打开用以后续分析的pcatmp.eigenvec查看一下
AB AB 0.00131144 0.00396951 -0.0564857
BR-01 BR-01 0.0231107 0.0369411 -0.0218842
BR-03 BR-03 0.0193726 0.028917 0.0294029
BR-04 BR-04 0.0221467 0.0355209 0.0069615
BR-05 BR-05 0.0256852 0.0432579 0.0164723
BR-06 BR-06 0.0134884 0.0311958 -0.0547523
打开文本编辑器,将下面这一行加到分析文件的第一行中,以便后面的分析:
ID Group PC1 PC2 PC3
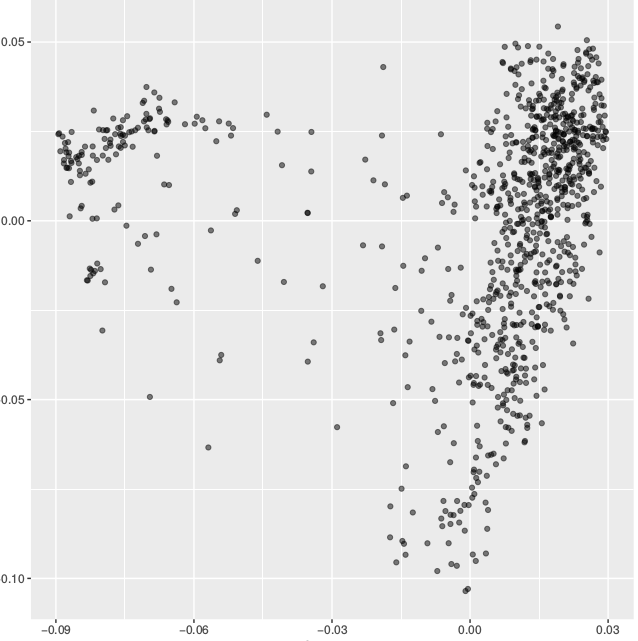
使用的R包进行可视化,这里没有具体将每个个体就行分群,我全部标记为一个组。真实的结果是根据你strcture图或者系统进化树对群体进行适当的分组,这样画出来的效果会更好,图例中PC1,PC2,PC3的值根据你生成的pcatmp.eigenval文件来计算。
library(ggplot2)
pdf(file="plot.evec1.pdf")
data <-read.table(file="pcatmp.eigenvec",header=T)
par(mfrow<-c(3,1))
ggplot(data = data, aes(x = PC1, y = PC2, group = Group)) +
geom_point(alpha = 0.5) + xlab("PC1(9.46%)") + ylab("PC2(3.23%)")
ggplot(data = data, aes(x = PC1, y = PC3, group = Group)) +
geom_point(alpha = 0.5) + xlab("PC1(9.46)") + ylab("PC3(2.43%)")
ggplot(data = data, aes(x = PC2, y = PC3, group = Group)) +
geom_point(alpha = 0.5) + xlab("PC2(3.23%)") + ylab("PC3(2.43%)")
dev.off()
实战二:EIGENSOFT中的smartpca
首先同样使用plink将vcf文件转格式转化成.ped和.map结尾的文件:
plink --allow-extra-chr --out myplink_test --recode --vcf Gpan.prune.in.vcf
进一步使用EIGENSOFT中内置的convertf 文件转化为smartpca的输入文件:
convertf -p transfer.conf
该步骤需要一个config file,将文件的输入输出写进去。然后执行command:
##config file
genotypename: myplink_test.ped
snpname: myplink_test.map # or example.map, either works
indivname: myplink_test.ped # or example.ped, either works
outputformat: EIGENSTRAT
genotypeoutname: example.eigenstratgeno
snpoutname: example.snp
indivoutname: example.ind
familynames: NO
该步骤会生产生三个pca所需的输入文件 example.eigenstrat example.snp example.ind
smartpca -p runningpca.conf
其config file 如下,根据你的数据参照manual来修改对应的参数:
genotypename: example.geno
snpname: example.snp
indivname: example.ind
evecoutname: example.pca.evec
evaloutname: example.eval
altnormstyle: NO
numoutevec: 10
numoutlieriter: 5
outliersigmathresh: 6.0
截取了运行中的屏幕输出,
number of samples used: 955 number of snps used: 338985
Using 47 threads, and partial sum lookup algorithm.
snp 01_8639 ignored . allelecnt: 0 missing: 155
snp 01_374042 ignored . allelecnt: 0 missing: 110
snp 01_519924 ignored . allelecnt: 0 missing: 202
snp 01_849970 ignored . allelecnt: 0 missing: 116
snp 01_987052 ignored . allelecnt: 0 missing: 120
snp 01_1320103 ignored . allelecnt: 0 missing: 233
snp 01_1321733 ignored . allelecnt: 0 missing: 60
snp 01_1414591 ignored . allelecnt: 0 missing: 144
snp 01_1630546 ignored . allelecnt: 0 missing: 107
snp 01_1659746 ignored . allelecnt: 0 missing: 185
total number of snps killed in pass: 7107 used: 331878
REMOVED outlier SRR1533159 iter 1 evec 9 sigmage 6.238 pop: Control
REMOVED outlier SRR1533177 iter 1 evec 9 sigmage 6.238 pop: Control
REMOVED outlier SRR1533192 iter 1 evec 9 sigmage 6.098 pop: Control
可以看到,smartpca有对数据进行统计与过滤处理,这里有一些地质量的snp和三个样本个体被去除。生成的结果文件如下,截取第2,3,4列,填充表头 ID Group PC1 PC2 PC3, 然后使用上面的Rscript进行可视化
#eigvals: 90.057 30.783 23.151 20.587 15.880 11.750 10.447 9.063 8.713 7.348
AB -0.0010 0.0046 0.0572 -0.0365 -0.0347 0.0246 0.0058 -0.0259 -0.0424 0.0130 Control
BR-01 -0.0237 0.0373 0.0228 0.0190 -0.0503 -0.0666 0.0262 0.0426 0.0098 0.0347 Control
BR-03 -0.0198 0.0293 -0.0287 0.0074 -0.0432 0.0582 -0.0310 0.0031 0.0313 -0.0240 Control
BR-04 -0.0228 0.0360 -0.0062 0.0265 -0.0686 -0.0364 0.0007 0.0340 -0.0091 -0.0015 Control
BR-05 -0.0265 0.0438 -0.0157 0.0262 -0.0468 -0.0147 -0.0185 0.0372 -0.0143 0.0350
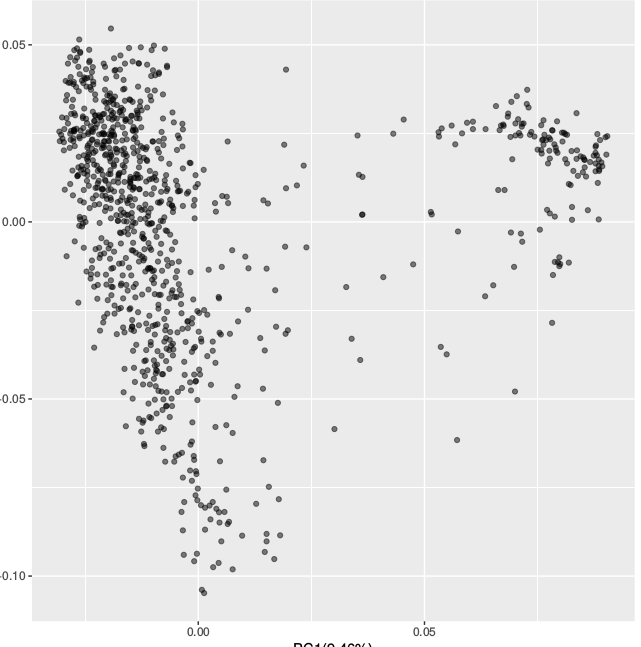
对比两个工具结果,维度不同,但cluster的结果还是很接近的。
实战3:最近出的R包工具
这个实战不同于上面,使用的是另一个数据
Load 对应的R包
library(vcfR)
library(poppr)
library(ape)
library(RColorBrewer)
读取vcf文件,还有数据信息文件,将vcf转化成genlight格式
rubi.VCF <- read.vcfR("prubi_gbs.vcf")
rubi.VCF
pop.data <- read.table("population_data.gbs.txt", sep = "\t", header = TRUE)
pop(gl.rubi) <- pop.data$State
gl.rubi <- vcfR2genlight(rubi.VCF)
使用glPCA function进行PCA分析
rubi.pca <- glPca(gl.rubi, nf = 3)
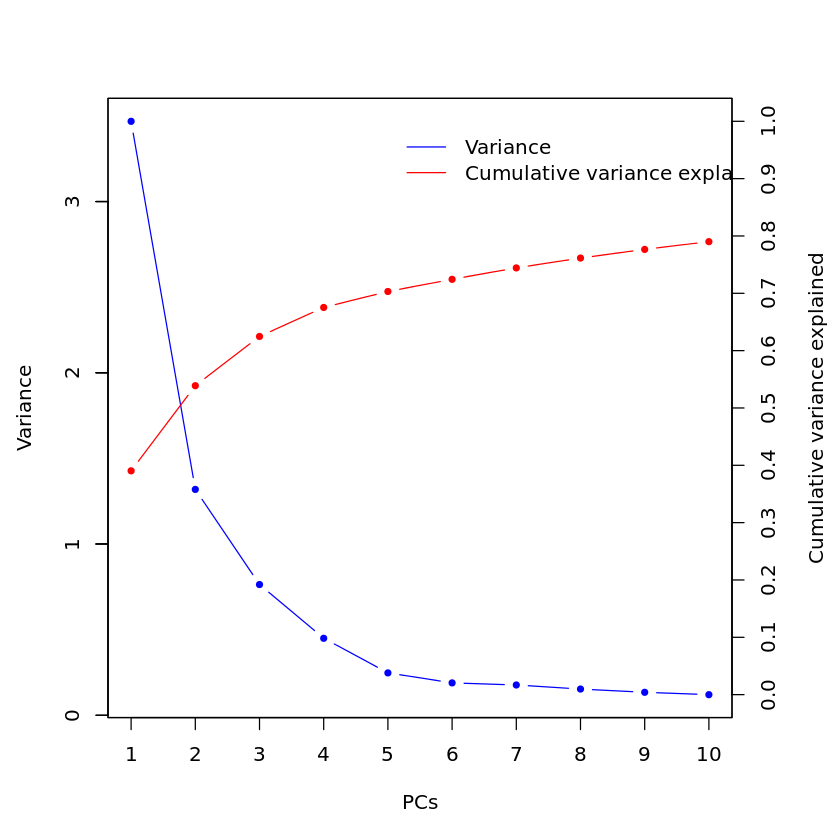
分析完后,对特征值用图形进行大概了解。
variance <- as.matrix(rubi.pca$eig)
par(mar = c(5.1,4.5,4.1,4.5))
plot(variance[1:10], type = "b", col = "blue", pch = 20, xaxt="n",yaxt="n", xlab="PCs",ylab="Variance")
axis(side = 1, at = 1:10)
axis(side = 2, at = round(seq(0, max(variance), length = 12)))
ec <- vector(mode="numeric", length=length(variance))
for (i in 1:length(variance)){
ec[i] <- sum(variance[1:i])/sum(variance)
}
par(new=TRUE)
plot(ec[1:10], type="b", col="red",xaxt="n",yaxt="n",xlab="",ylab="", ylim = c(0,1), pch = 20)
axis(side = 4, at = seq(0, 1, by = 0.1))
mtext("Cumulative variance explained", side=4, line=3)
legend(x = 5, y = 1.0, bty = "n",col=c("blue", "red"),lty=1,legend=c("Variance","Cumulative variance explained"))
结果如下图:前3个PCA累计解释63.183%的数据差异。
 2.png-42.6kB
2.png-42.6kB
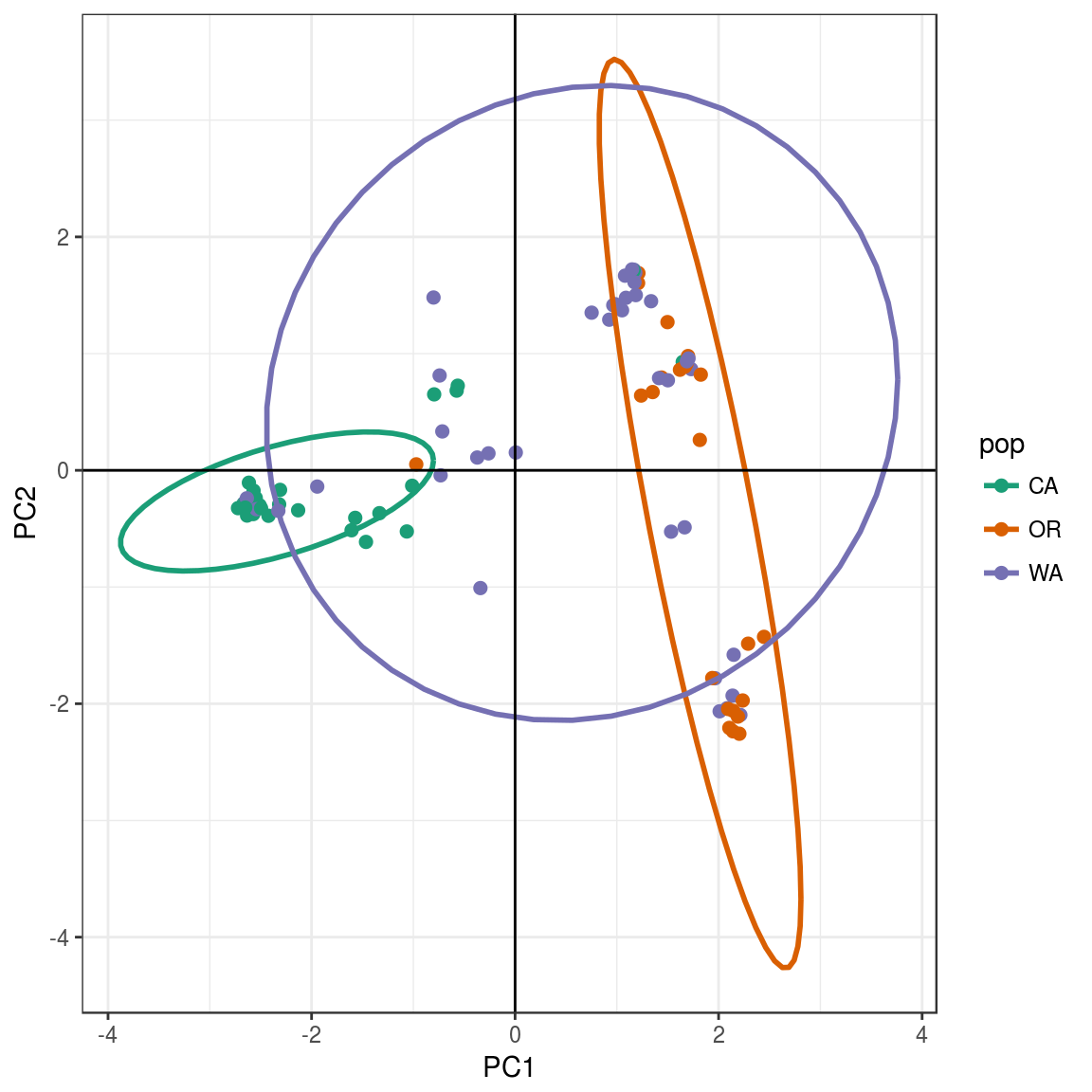
要查看PCA的结果,我们可以使用R包ggplot2。我们需要将包含主要组件(rubi.pca $scores)的数据框转换为新的对象rubi.pca.scores。另外,我们将在rubi.pca.scores对象中添加人口值作为新列,以便能够按种群对样本着色。ggplot2将绘制PCA图,根据总体对样本着色,并创建包含每个群体95%数据的椭圆圈。
rubi.pca.scores <- as.data.frame(rubi.pca$scores)
pop(gl.rubi) <- pop.data$State
rubi.pca.scores$pop <- pop(gl.rubi)
cols <- brewer.pal(n = nPop(gl.rubi), name = "Dark2")
set.seed(9)
p <- ggplot(rubi.pca.scores, aes(x=PC1, y=PC2, colour=pop))
p <- p + geom_point(size=2)
p <- p + stat_ellipse(level = 0.95, size = 1)
p <- p + scale_color_manual(values = cols)
p <- p + geom_hline(yintercept = 0)
p <- p + geom_vline(xintercept = 0)
p <- p + theme_bw()
p
结果如下:PCA分析将WA和CA样本(左边)还有样本CA和WA(右边)分离开来。然后样本CA比较显著的聚集在一起。
 3.png-95.3kB
3.png-95.3kB
PCA应用
-
群体分层分析和推断进化关系
可以准确的展示群体分层的类型,一般可以与phylogentic tree,structure,admixture的结果互相验证。
如图左边绿色是野生的大麦,右边橙色是驯化的大麦,中间蓝色是admixture混合的大麦。不同品系的大麦可以看到明显的分层,并且具有野生和驯化大麦特征的混合小麦位于野生还有驯化小麦之间。很好的反映了,混合小麦是其两者的一种中间过度形态,具有两者的特征。
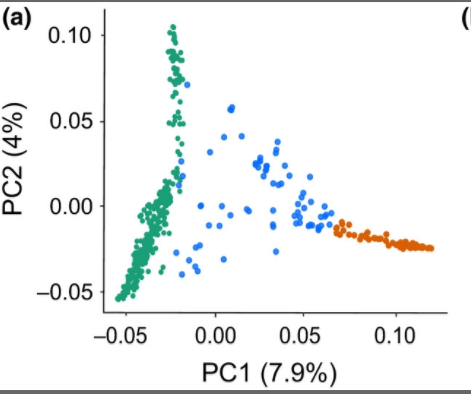 1.PNG-74.9kB
1.PNG-74.9kB
2.检查outliner
在一个群体中,如果其中有一些样本取样错误,或者测序时候有严重的污染,这样往往会导致outliner的产生,如下图,右上方的那个样本就是明显的outlier,有着与其他样本不同,偏高的PCA比值。当确认outliner后,可以准确校正群体的样本,减少对后续关联分析的影响。
 此处输入图片的描述
此处输入图片的描述










网友评论
Error: Line 1 of [tmp.bim] contains illegal chr number, please check
An error occurs, please check the options or data
KZ288185.1 . 0 125 T C就是tmp.bim的首行。恳请老师指导如何解决。