RNA-seq工具哪家强
RNA-seq分析工具知多少
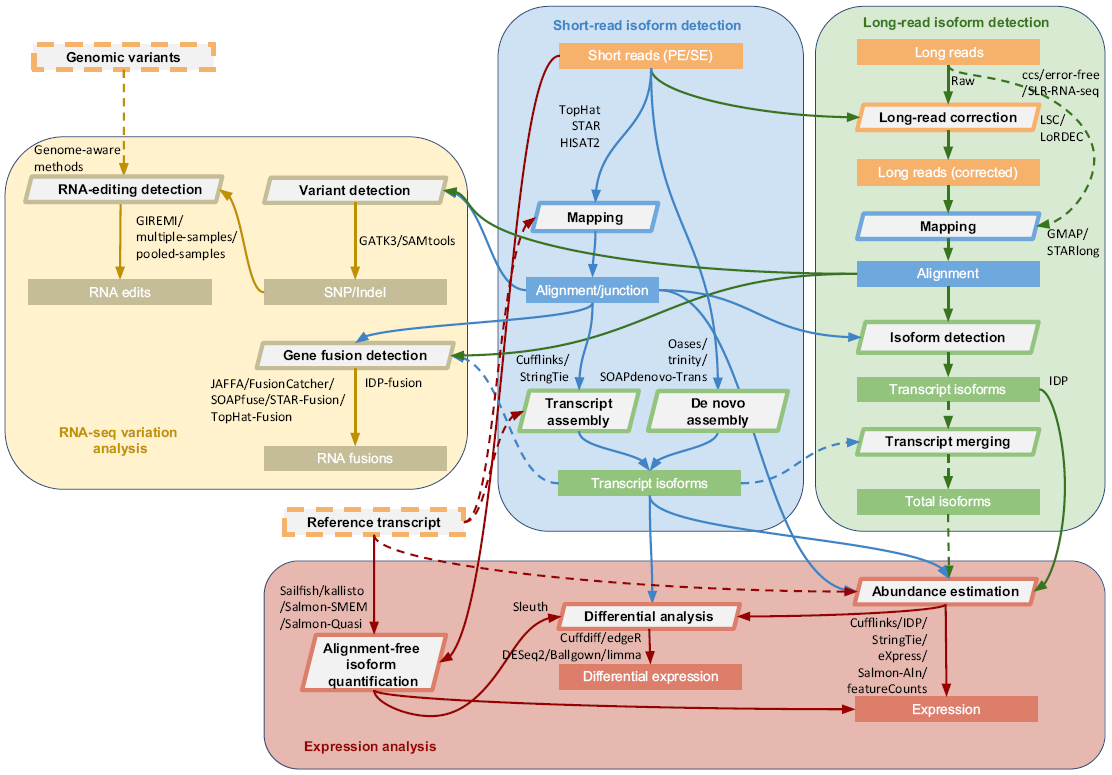
RNA-seq是研究转录组应用最广泛,也最重要的技术之一。RNAseq其分析内容包括序列比对、转录本拼装、表达定量、差异分析、融合基因检测、可变剪接、RNA编辑和突变检测等,具体流程和常用工具如下图所示。通常的分析不一定需要走完全部流程,按需进行,某些步骤可以跳过、简化等。
RNA-seq分析工具最优组合
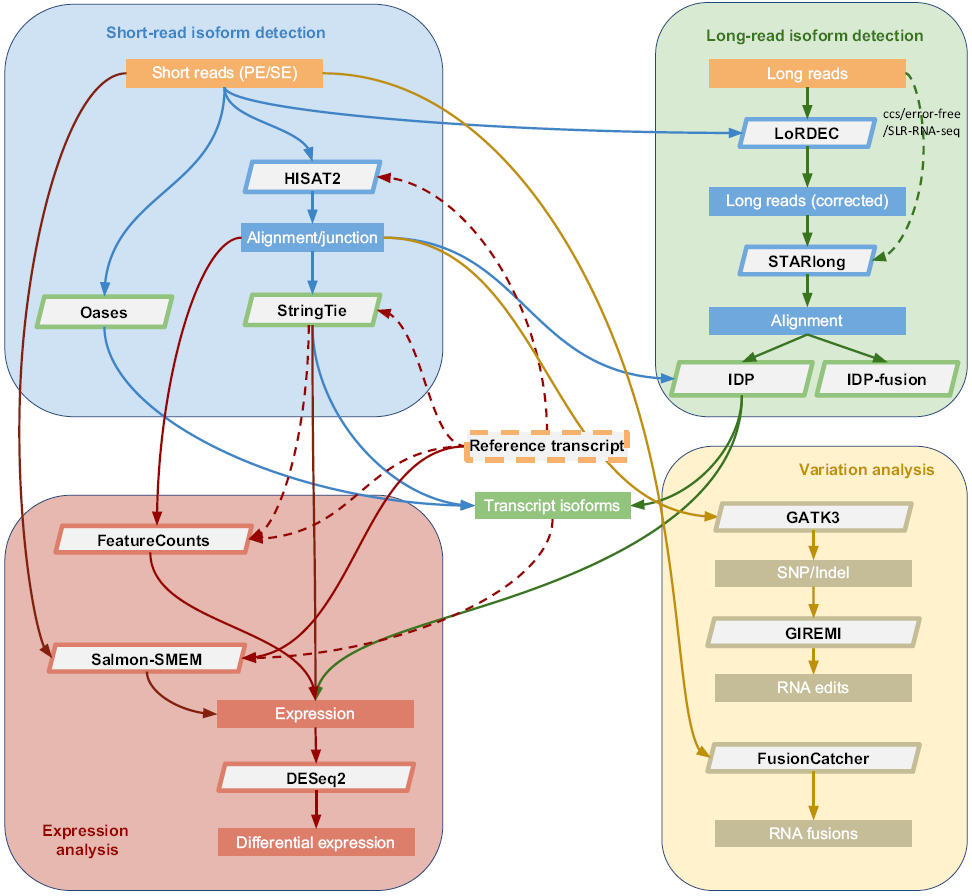
Nature Communication上一篇文章Gaining comprehensive biological insight into the transcriptome by
performing a broad-spectrum RNA-seq analysis对15个样品(正常样品、癌细胞和干细胞,短读长和长读长)的转录组数据利用39个分析工具,120种常见组合方式进行的490次深入分析,并以测序质量控制联盟(SEQC)的qPCR检测结果做为正对照,总结出一套普适性流程,如下。
通过综合分析RNA-seq分析流程中不同步骤的工具性能发现不同的分析工具和方法对分析结果的准确度和分析时间影响巨大。HISAT2表现出最快的速度和最准确的拼接比对,但是没有STAR的敏感度高。StringTie在速度和准确度上都优于Cufflinks。
长读段方法如IDP和Iso-Seq会识别许多短读段技术没有识别到的多外显子转录本,但是会丢失一些单外显子转录本。
不经过比对的工具如Salmon-SMEM和kallisto获得了最好的一致性和最高准确度,因此,如果目标不是发现新的转录本,如Salmon-SMEM和kallisto可以作为准确而快速的解决方案。DESeq2和edgeR与不经过比对的工具联用可以获得高准确度的差异表达分析结果。
通常情况下,整体最好的分析流程对于特定的数据集特定的研究目的来说可能是次优的。比如,对于比对和转录组构建,HISAT2-StringTie组合具有更高的准确度和更快的速度。但是对于MCF7-300样品来讲,STAR- StringTie组合具有更高的灵敏度。
序列比对质量大比拼
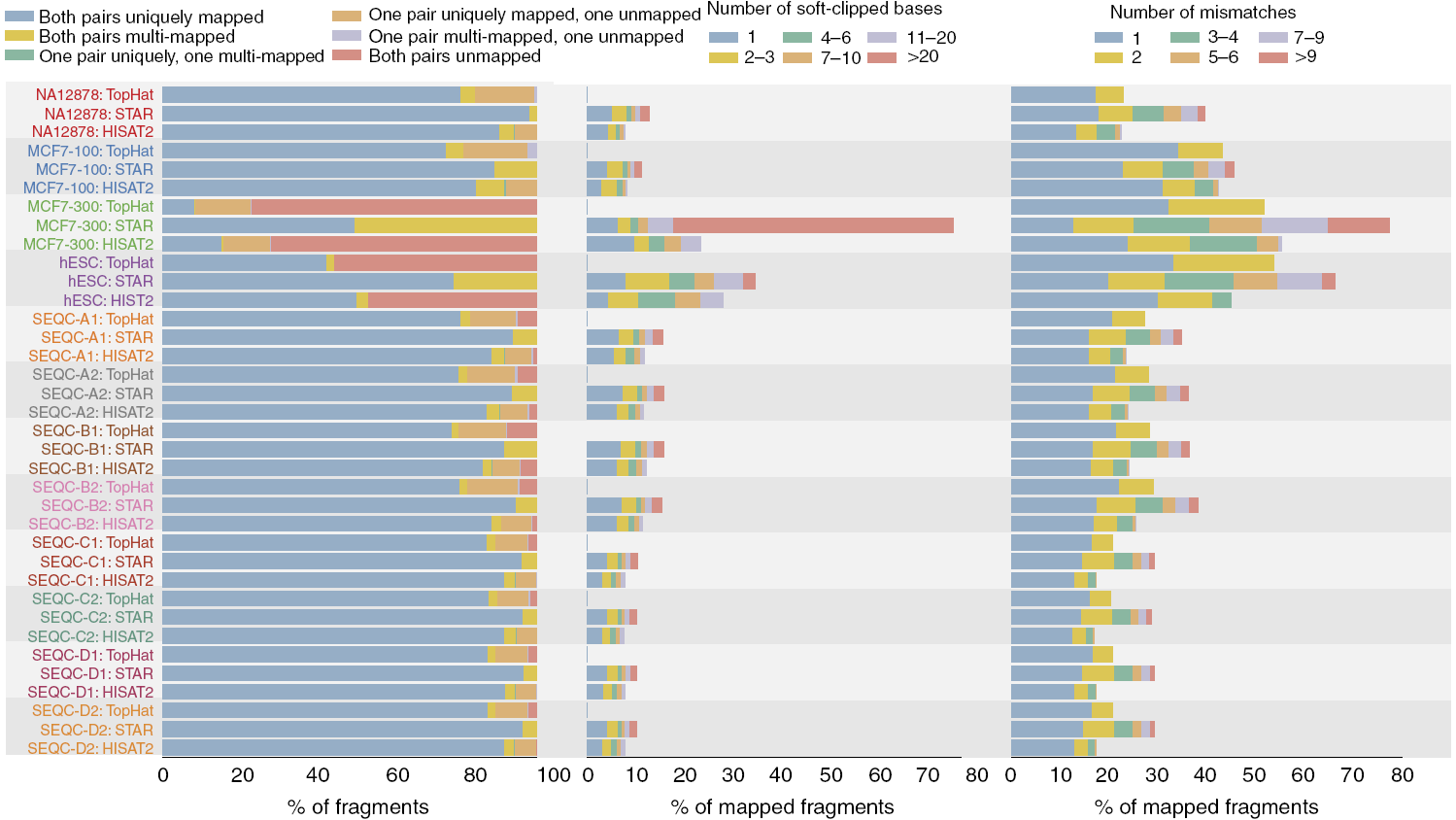
STAR具有最高比例的在基因组上有唯一比对位置的reads,尤其是对读长为300 nt的MCF7样品也有最高的比对率。
与TopHat和HISAT2不同,STAR只保留双端reads都比对到基因组的序列,但对低质量的比对(允许更多的错配碱基和soft-clip事件)容忍度高。这一点在长reads (MCF7-300)样品中的体现更为明显。TopHat则不允许soft-clip事件。soft-clip事件:即reads末端存在低质量碱基或接头导致比对不上的, STAR会自动尝试截去未比对部分,只保留比对上的部分。
在比对速度方面,HISAT2比STAR快2.5倍,比TopHat快大约100倍。
(后续会推出柱状图的一步画法)
Exon-exon junction位点评估
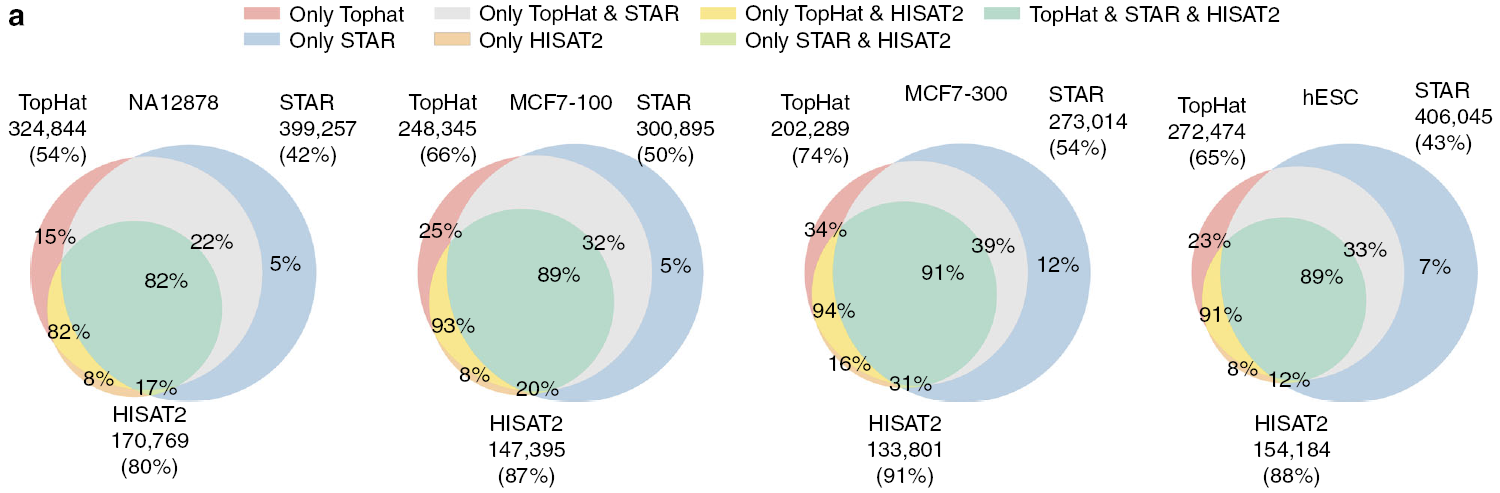
转录组reads比对不同于基因组reads比对(如ChIP-seq、WES等)的地方在于比对的reads可能来源于2个被内含子隔开的外显子区域,导致reads一端比对在第一个外显子的后面部分,另一端比对在第二个外显子的前面部分,从而形成exon-exon
junction (剪接点)。这些reads又称为junction reads,对转录本的拼接、鉴定和差异分析具有重要的意义。
下面的维恩图展示了不同比对软件检测到的共有和特有的剪接位点的比较(整数代表每个软件检测到的剪接位点的数目,百分数代表每个集合的splice junction被验证的比例)。可信的剪接点定义为dbEST数据库中有至少2个表达序列标签(EST)支持的位点,做为正对照。HISAT2在所有样品中拥有最高的剪接点验证率(80%-91%),TopHat其次(54%-74%),STAR最低(42%-54%)。但是HISAT2预测的剪接点的数量最少,约为TopHat的60%和STAR的50%。
韦恩图绘制看R语言学习-韦恩图轻松绘制各种Venn图
基于参考基因组的转录组组装
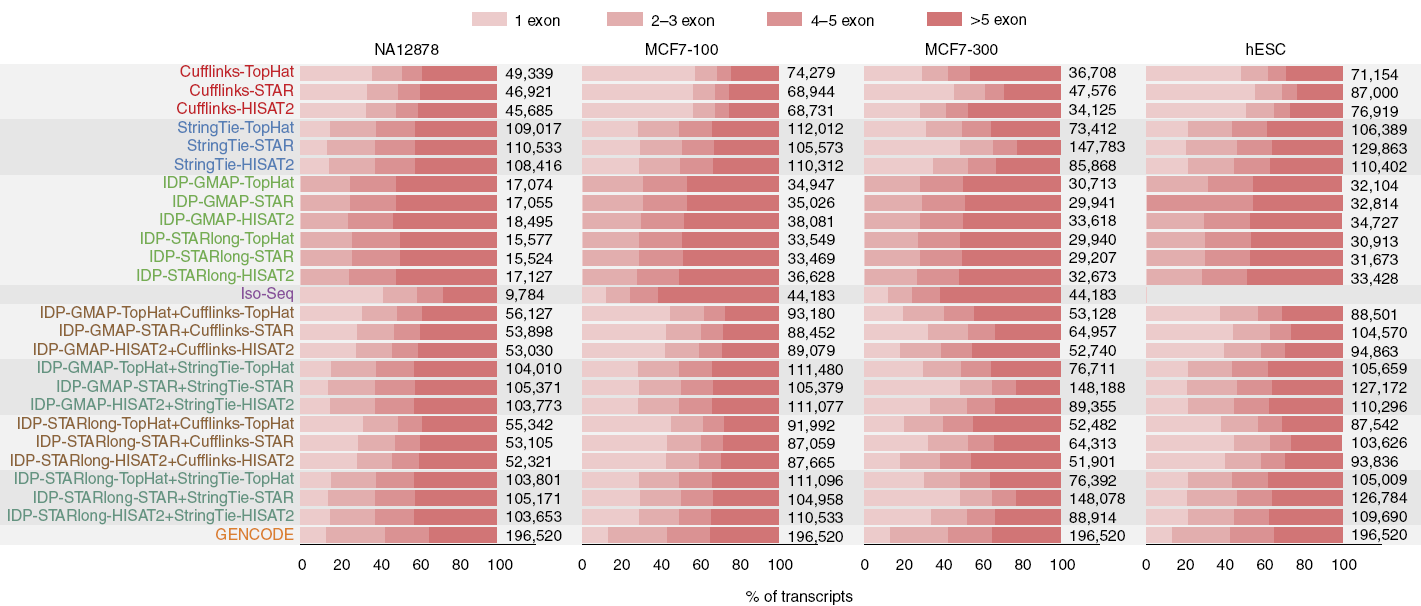
对于二代测序数据,Cufflinks和StringTie是应用最广泛的两个基于比对结果的转录本拼装工具。(比对软件STAR,HISAT2和TopHat)
对于三代测序数据,PacBio的流程中默认使用软件Iso-Seq。
二代和三代测序数据杂交拼装,使用的是IDP (Isoform Detection
and Prediction)。(比对软件GMAP、STAR long)
转录本拼装质量评估的依据是GENCODE v19的参考转录组注释,不存在于这个集合的转录本视为假阳性。
每个转录本中包含的外显子的数目是转录本拼装质量的一个评价标准,通常单外显子转录本可信度最差。Cufflinks的单外显子转录本的数目占到30%左右,StringTie在15%左右。这些单外显子转录本大约90%为假阳性(数字为目测附图的估计)。StringTie拼装获得的转录本的数目约为Cufflinks的两倍,其外显子数目的分布与GENCODE v19较为相似。IDP组装出的都是多外显子转录本,整体数目与Cufflinks排除单外显子转录本后相近,但外显子数目的分布与GENCODE
v19更一致。与之相比,Iso-Seq的假阳性率较高,但敏感性更强。
堆积柱状图的画法将会后续推出。
对于基因水平的组装,IDP的的准确性和灵敏性都是最好的。Cufflinks比StringTie更为准确和灵敏。对于MCF3-300样品来讲,含有STAR的组合拼装出更多的转录本,但拼装准确性和灵敏性都略低于基于TopHat和HISAT2的结果。IDP和StringTie拼装出更多的多转录本基因。(下图左)
对于转录本水平的组装,IDP的准确性比其它技术高20%,但其敏感性低于StringTie,高于Cufflinks。相比喻Cufflinks,StringTie转录本水平的组装精确性和敏感性高11%和25%。在预测新的转录本上(ENSEMBL没有注释但GENCODE v19有的3681个转录本),StringTie得到的最多,约是Cufflinks和IDP的2.5和6.5倍。(下图右)
另外StringTie的速度是Cufflinks的50倍,IDP的60倍。
散点图绘制R语言学习-散点图绘制
表达定量
传统的表达分析是将reads比对回参考基因组或者参考转录组,然后估计转录本丰度。如果研究目的是关注已知的和新的转录本的丰度,比对回参考基因组后使用Cufflinks和StringTie进行组装,然后评估表达丰度。如果只想定量已经注释的基因,直接比对到参考转录组,再使用RSEM和eXpress进行丰度估计。
现在基于转录本的定量还有一种方式是不经过比对直接判断read来源于哪个转录本,这比拼接比对定量需要更少的计算资源。Sailfish、Salmon、quasi-mapping和kallisto四种工具是这一计算方式的代表。
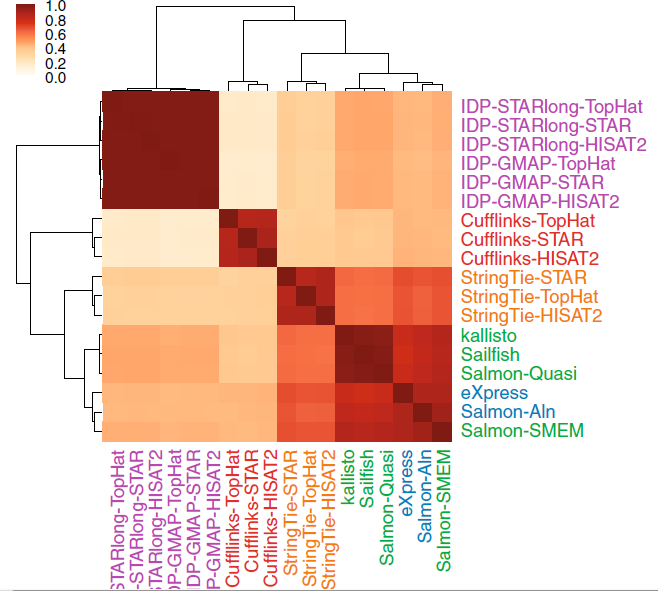
对样品NA12878采用不同方法定量得到的基因表达谱进行log转换后的Spearman秩和相关性分析表明采用相似方法的定量工具获得的表达图谱更相近。Cufflinks的定量结果与其他工具相关性最差,不足0.4.不需要比对直接定量的工具与StringTie计算的结果更相近(相关系数0.6-0.8)。Salmon-SMEM与基于转录组比对的工具eXpress和Salmon-Aln聚在一起,但Salmon-SMEM运行速度更快。
R语言学习-热图简化
R语言学习-热图美化
R语言学习-热图绘制(heatmap)
对于同一个样品不同测序读长的数据(MCF7-100和MCF7-300)的比较分析可以反应比对工具定量的稳定性。两个不依赖于比对的定量工具kallisto和Salmon-SMEM具有最一致的定量结果。Cufflinks-TopHat组合的结果在基于比对的定量工具组合中表现最优。整体看,基于STAR的比对结果,定量稳定性低于基于HISAT2的比对。
综上,不基于比对的定量结果效率和稳定性最高。StringTie与HISAT2的组合是基于比对的定量工具中性能最好的,但也要比不基于比对的工具慢一个数量级。
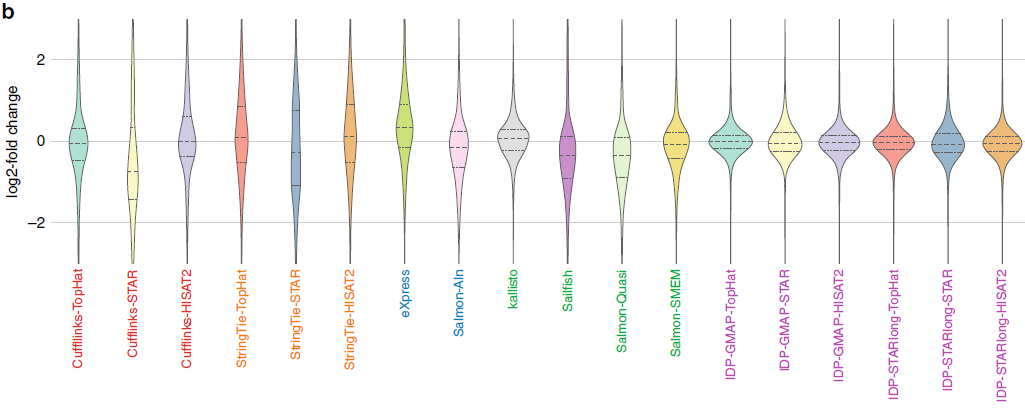
此图为小提琴图(R语言学习-箱线图(小提琴图、抖动图、区域散点图)
R语言学习-箱线图一步法),展示了数据分布的密度,越胖的地方数据越集中。纵向表示两个样品基因表达变化的幅度,横向表示变化幅度的集中度,数据越集中于y=0,定量一致性越好。
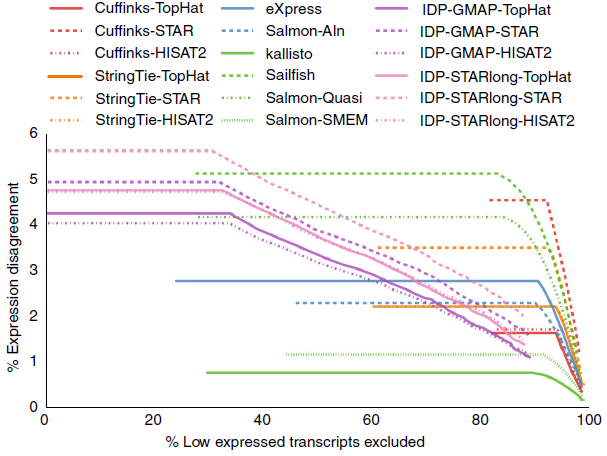
此图为线图(R语言学习-线图一步法
R语言学习-线图绘制),展示的是逐步移除最低表达的部分转录本后定量的一致性。线越接近X轴表明一致性越好。
差异表达基因鉴定
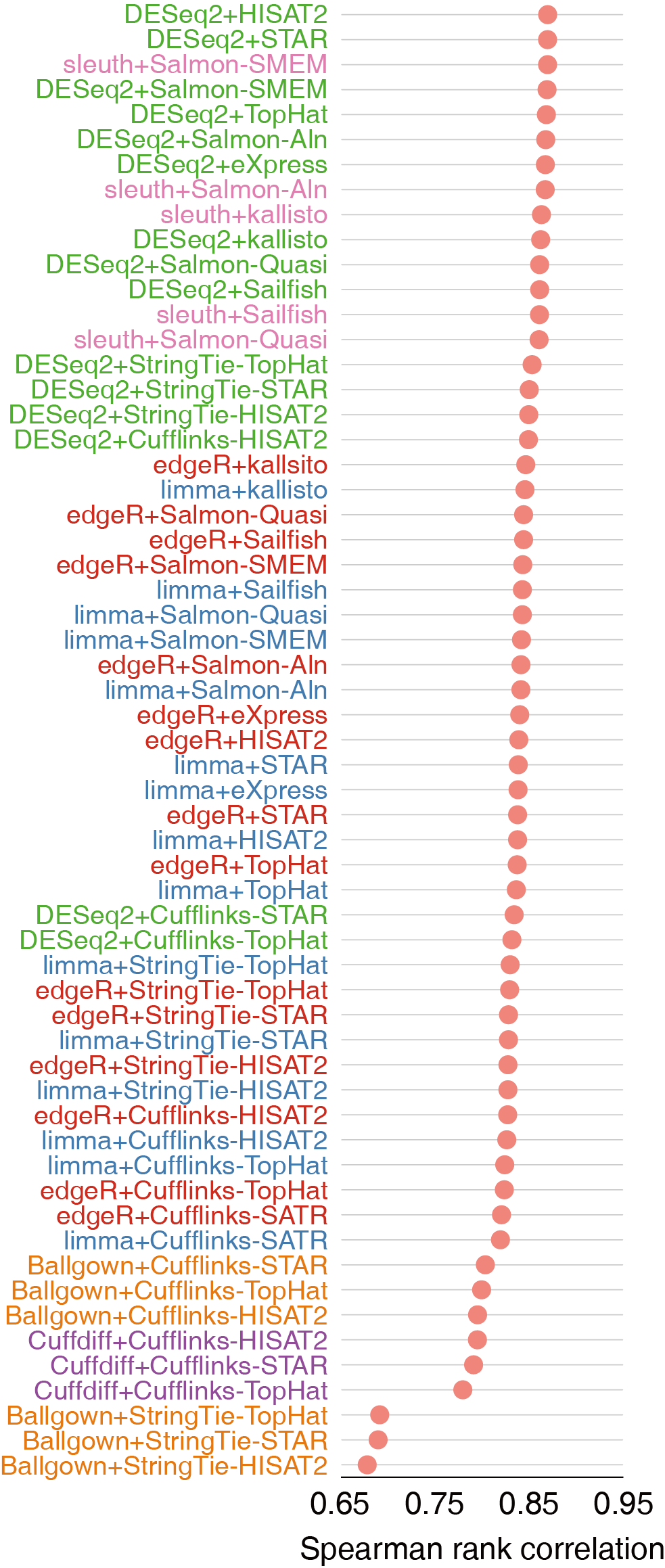
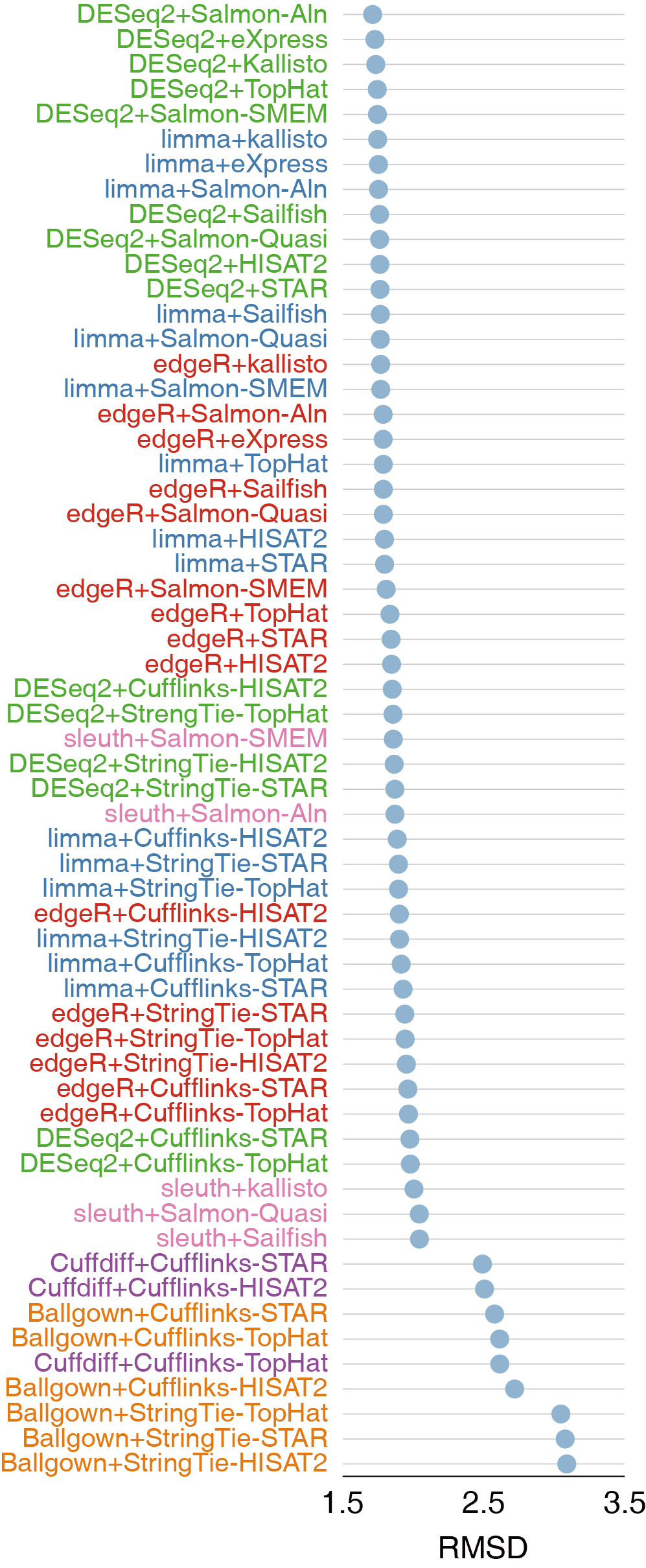

不同样品和条件下差异表达基因的识别是RNA-seq分析的重要目标。有多种方法鉴定差异表达基因,包括基于计数(reads count)的DESeq、limma和edgeR、基于组装技术的Cuffdiff和Ballgown、不经过比对定量进行差异分析的sleuth。
SEQC样品(SEQC-A vs
SEQC-B, SEQC-C vs SEQC-D)中1001个有qRT-PCR定量过的基因作为对照评价工具的性能。DESeq2在所有组合中表现最佳,sleuth、edgeR和limma略微次之,但差别不大。Cuffdiff和Ballgown的准确度没有基于计数的工具准确度高。
对于AUC-30的估计,edgeR表现最佳, DESeq2与之差别不大。
基于来讲基于计数的工具比基于组装的工具更高效,不经过比对直接定量的工具如Salmon和kallisto能够获得高质量的差异分析结果。
以上三个图都是散点图,第一个Spearman rank correlation相关性越高越好,第二个RMSD类似于均方差(与对照相比得分偏差的平方和先求均值再开方),第三个AUC-30表示在假阳性率为30%时ROC曲线下的面积,面积越大表示结果越准确(纵轴是True positive rate)。
加入生信宝典,一起换个角度学生信
http://mp.weixin.qq.com/s/NUEi6oRFL7B3f1FpCD4Xug
http://mp.weixin.qq.com/s/xAaj-d5LRRj0SSMFJ7Yo9Q
联系我们





网友评论