tags: HISAT2 RNA-seq
HISAT2 发表的文章中强调了它的速度很快,我就测试了一下这个工具。
HISAT2 建立索引:
hisat2-build -p 4 rRNA.fa rRNA.fa.tran
然而没多久就看到这样的警告:
Reading reference sizes
Warning: Encountered reference sequence with only gaps
Warning: Encountered reference sequence with only gaps
Warning: Encountered reference sequence with only gaps
Warning: Encountered reference sequence with only gaps
Warning: Encountered reference sequence with only gaps
只是警告,并没有报错。
HISAT2 建参考索引很慢,等 HISAT2 建完索引(建索引花了 16 个小时),然后用 HISAT2 比对 RNA-seq 数据测试。
hisat2 -S /dev/null -p 4 -x rRNA.fa.tran -1 A_1.fq.gz -2 A_2.fq.gz --un-conc-gz A_filter_rRNA_%.fq.gz
几分钟之后报错了:
(ERR): hisat2-align died with signal 11 (SEGV) (core dumped)
github 上有人在 HISAT2 项目中报告过这个错误,虽然没有最终讨论出解决办法,但是都觉得跟建索引不完整有关,或许与建索引时候的警告有关。我查了一些资料,综合 biostar 和 SEQanswer 中的讨论,建立索引时遇到的警告是由于参考序列中存在大段的 n 碱基导致的,例如其中一条 fasta 中 n (我遇到的是小写的 n)太多。解决办法也很简单,过滤掉参考序列中长度小于 50bp 的 contig 和序列中连续 n 碱基超过 40bp 的contig 。然后重新建索引,就没有任何警告了,但是会损失部分参考序列。这样处理之后建的索引再用 HISAT2 比对 RNA-seq 数据,就没有问题了。
#!/usr/bin/env python3
from Bio import SeqIO
long_seq = []
for record in SeqIO.parse("rRNA.fa","fasta"):
if len(record.seq) > 50 and not 'n'*40 in record.seq:
long_seq.append(record)
SeqIO.write(long_seq, "filtered.rRNA.fa", "fasta")
HISAT2 比对速度确实很快,一个样本转录组数据比对 2G 的核糖体 RNA 参考基因组约 25 分钟,bowtie2 需要 170 分钟(线程数都是 4)。 bowtie2 的 local 模式比对出 21% 的 rRNA 污染,而 HISAT2 比对 2% 的 rRNA 污染,差异也挺大的……
但是,HISAT2 的线程数不能设置太高,用户手册中建议对人类参考基因组进行比对时,线程数设置在 1-8 之间。我用文献 Transcript-level expression analysis of RNA-seq
experiments with HISAT, StringTie and Ballgown 中的数据测试也发现当线程数超过 12 时整个比对步骤消耗的时间反而会增加。
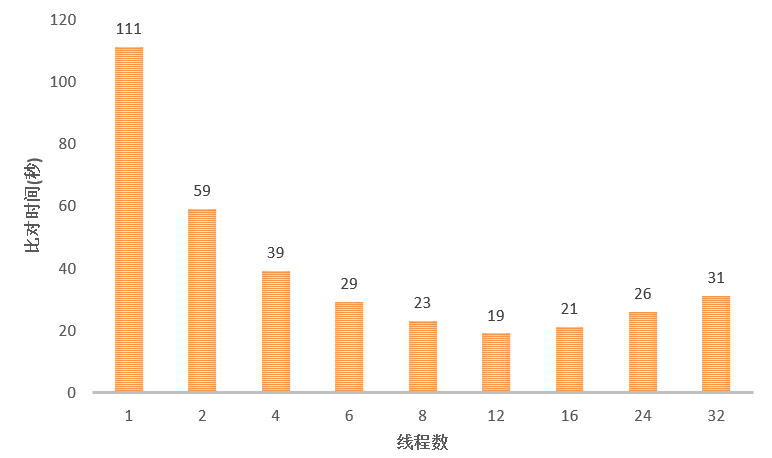 HISAT2 比对线程数测试
HISAT2 比对线程数测试




网友评论