tags: RNA-seq GATK STAR SNP INDEL
RNA-seq 序列比对
对 RNA-seq 产出的数据进行变异检测分析,与常规重测序的主要区别就在序列比对这一步,因为 RNA-seq 的数据是来自转录本的,比对到参考基因组需要跨越转录剪切位点,所以 RNA-seq 进行变异检测的重点就在于跨剪切位点的精确序列比对。
有一篇文献 systematic evaluation of spliced alignment programs
for RNA-seq data 对 RNA-seq 数据常用的 11 款比对软件进行了详细的比较测试,例如 tophat2, STAR 等。 GATK 发布的 RNA-seq 数据变异检测最佳实践流程用了 STAR 2-pass 这一方法进行序列比对,STAR 发表的文章至今已被引用 1900 余次,这款软件的比对速度很快,也是 ENCODE 项目的御用比对软件。
STAR 2-pass 模式需要进行两次序列比对,建立两次参考基因组索引。它的思路是第一次建参考基因组索引之后进行初步的序列比对,根据初步比对结果得到该样本所有的剪切位点信息,包括参考基因组注释 GTF 中已知的剪切位点和比对时新发现的剪切位点,然后利用第一次比对得到的剪切位点信息重新对参考基因组建立索引,然后进行第二次的序列比对,这样可以得到更精确的比对结果。
这里使用了一个测试数据演示流程,第一次对参考基因组建索引:
# star 1-pass index
STAR --runThreadN 8 --runMode genomeGenerate \
--genomeDir ./star_index/ \
--genomeFastaFiles ./genome/chrX.fa \
--sjdbGTFfile ./genes/chrX.gtf
然后进行第一次序列比对:
#star 1-pass align
STAR --runThreadN 8 --genomeDir ./star_index/ \
--readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix ./star_1pass/ERR188044
之后根据第一次比对得到的所有剪切位点,重新对参考基因组建立索引:
# star 2-pass index
STAR --runThreadN 8 --runMode genomeGenerate \
--genomeDir ./star_index_2pass/ \
--genomeFastaFiles ./genome/chrX.fa \
--sjdbFileChrStartEnd ./star_1pass/ERR188044SJ.out.tab
再进行 STAR 二次序列比对:
# star 2-pass align
STAR --runThreadN 8 --genomeDir ./star_index_2pass/ \
--readFilesIn ./samples/ERR188044_chrX_1.fastq.gz ./samples/ERR188044_chrX_2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix ./star_2pass/ERR188044
由于后面要用 GATK 进行 call 变异,还需要对比对结果 SAM 文件进行一些处理,这些都可以用 picard 来做,包括 SAM 头文件添加 @RG 标签,SAM 文件排序并转 BAM 格式,然后标记 duplicate:
# picard Add read groups, sort, mark duplicates, and create index
java -jar picard.jar AddOrReplaceReadGroups \
I=./star_2pass/ERR188044Aligned.out.sam \
O=./star_2pass/ERR188044_rg_added_sorted.bam \
SO=coordinate \
RGID=ERR188044 \
RGLB=rna \
RGPL=illumina \
RGPU=hiseq \
RGSM=ERR188044
java -jar picard.jar MarkDuplicates \
I=./star_2pass/ERR188044_rg_added_sorted.bam \
O=./star_2pass/ERR188044_dedup.bam \
CREATE_INDEX=true \
VALIDATION_STRINGENCY=SILENT \
M=./star_2pass/ERR188044_dedup.metrics
到此序列比对就完成了。
使用 GATK 进行变异检测
感觉 GATK 里面的工具都很慢(相对于其他的软件特别慢!),都是单线程在跑,有的虽然可以设置为多线程但是感觉没啥速度上的提升,所以要有点耐心……
由于 STAR 软件使用的 MAPQ 标准与 GATK 不同,而且比对时会有 reads 的片段落到内含子区间,需要进行一步 MAPQ 同步和 reads 剪切,使用 GATK 专为 RNA-seq 应用开发的工具 SplitNCigarReads 进行操纵,它会将落在内含子区间的 reads 片段直接切除,并对 MAPQ 进行调整。DNA 测序的重测序应用中也有序列比对软件的 MAPQ 与 GATK 无法直接对接的情况,需要进行调整。
# samtools faidx chrX.fa
# samtools dict chrX.fa
java -jar GenomeAnalysisTK.jar -T SplitNCigarReads \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup.bam \
-o ./star_2pass/ERR188044_dedup_split.bam \
-rf ReassignOneMappingQuality \
-RMQF 255 \
-RMQT 60 \
-U ALLOW_N_CIGAR_READS
之后就是可选的 Indel Realignment,对已知的 indel 区域附近的 reads 重新比对,可以稍微提高 indel 检测的真阳性率,如果时间紧张也可以不做,这一步影响很轻微 :)
# 可选步骤 IndelRealign
java -jar GenomeAnalysisTK.jar -T RealignerTargetCreator \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-o ./star_2pass/ERR188044_realign_interval.list \
-known Mills_and_1000G_gold_standard.indels.hg19.sites.vcf
java -jar GenomeAnalysisTK.jar -T IndelRealigner \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-known Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-o ./star_2pass/ERR188044_realign.bam \
-targetIntervals ./star_2pass/ERR188044_realign_interval.list
然后还是可选的 BQSR,这一步操作主要是针对测序质量不太好的数据,进行碱基质量再校准,如果对自己的测序数据质量足够自信可以省略,2500 之后 Hiseq 仪器的数据质量都挺不错的,可以根据 FastQC 结果来决定。这一步省了又能节省时间 :)
# 可选步骤 BQSR
java -jar GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_realign.bam \
-knownSites 1000G_phase1.snps.high_confidence.hg19.sites.vcf \
-knownSites Mills_and_1000G_gold_standard.indels.hg19.sites.vcf \
-o ./star_2pass/ERR188044_recal_data.table
java -jar GenomeAnalysisTK.jar \
-T PrintReads \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_realign.bam \
-BQSR ./star_2pass/ERR188044_recal_data.table \
-o ./star_2pass/ERR188044_BQSR.bam
比如下面的数据就可以放心的省略这两步了:
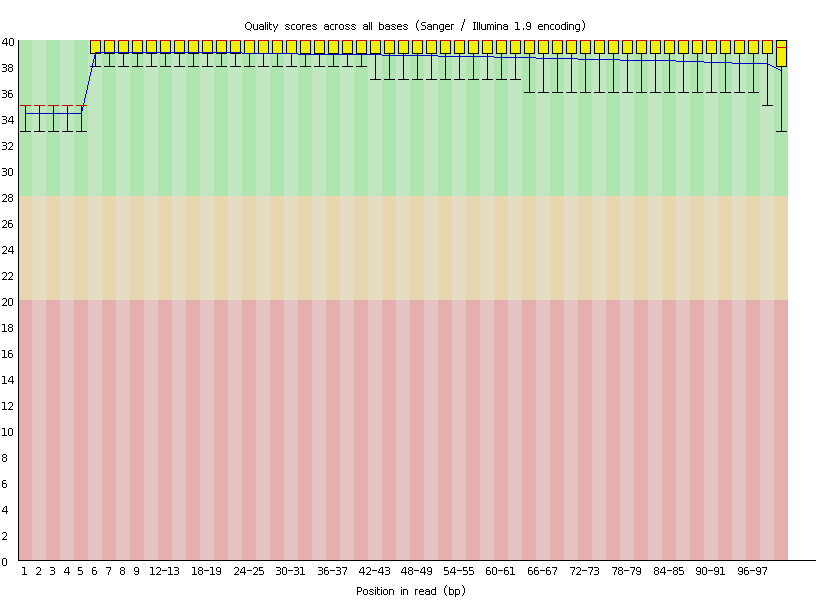 R1 数据质量够好
R1 数据质量够好
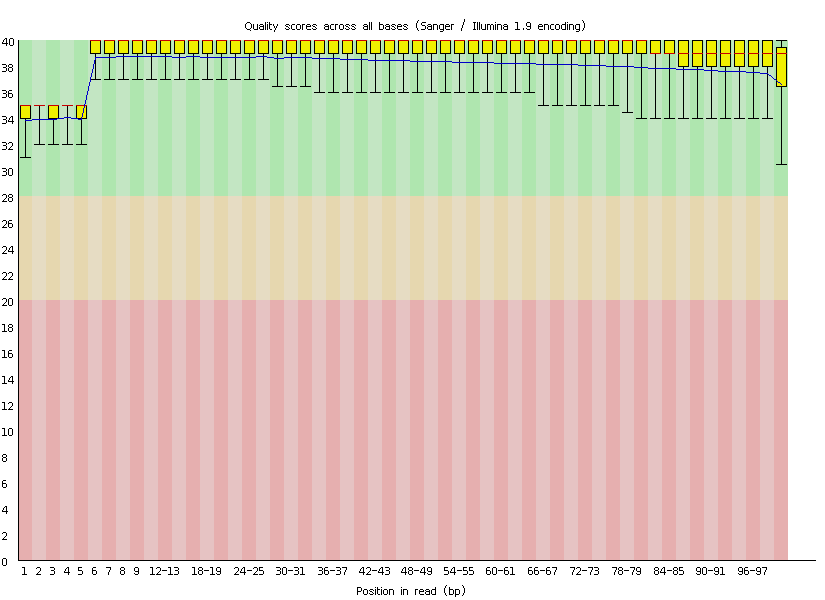 R2 数据质量也够好
R2 数据质量也够好
现在终于可以进行变异检测了,GATK 官网说 HC 表现比 UC 好,所以这里用 HC 进行变异检测:
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R ./genome/chrX.fa \
-I ./star_2pass/ERR188044_dedup_split.bam \
-dontUseSoftClippedBases \
-stand_call_conf 20.0 \
-o ./star_2pass/ERR188044.vcf
call 完变异之后再进行过滤:
java -jar GenomeAnalysisTK.jar \
-T VariantFiltration \
-R ./genome/chrX.fa \
-V ./star_2pass/ERR188044.vcf \
-window 35 \
-cluster 3 \
-filterName FS -filter "FS > 30.0" \
-filterName QD -filter "QD < 2.0" \
-o ./star_2pass/ERR188044_filtered.vcf
然后就拿到变异检测结果了,可以用 ANNOVAR 或 SnpEff 或 VEP 进行注释,根据自己的需要进行筛选了。




网友评论