最近要处理一些基因注释的文件,要用到bedtools这款经典的软件,在慢慢接触到这款软件才发现其功能的强大之处,在接下来的几个星期内将会大家一起入门并且学习掌握使用bedtools。
关于bedtools的背景
bedtools的发展是由于需要快速,灵活的工具来比较大量的基因组特征。用现有工具回答基础研究问题要么太慢,要么需要修改他们报告或计算结果的方式,不方便生信工程师使用。基于这样的情况下,Bedtools一款用于g对enomicfeatures进行比较、相关操作和注释的工具在2009年被开发出来,目前版本已经有三十多个工具/命令用以实现各种不同的功能,可以针对bed、vcf、gff等格式的文件进行处理。
工具下载
通过Github从源代码编译稳定版本的bedtools.
wget https://github.com/arq5x/bedtools2/releases/download/v2.25.0/bedtools-2.25.0.tar.gz
tar -zxvf bedtools-2.25.0.tar.gz
cd bedtools2
make
测试文件下载
建立,并进入新的文件夹
mkdir -p /scratch/pawsey0149/hhu/bio_trainee/bedtools/test_data
cd /scratch/pawsey0149/hhu/bio_trainee/bedtools/test_data
下载测试数据
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/maurano.dnaseI.tgz
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/cpg.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/exons.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/gwas.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/genome.txt
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/hesc.chromHmm.bed
解压压缩的数据
tar -zxvf maurano.dnaseI.tgz
rm maurano.dnaseI.tgz
简单看看这些测试数据是什么?
ls -1
这些文件中的20个(以f开头的)是了在来自脑,心脏,肠,肾,肺,肌肉,皮肤和胃的20种不同胎儿组织样品中测量的DnaseI超敏感性位点。另外,
cpg.bed代表人类基因组中的CpG岛; exons.bed代表来自人类基因的RefSeq外显子; gwas.bed代表在全基因组关联研究(GWAS)中鉴定的人类疾病相关SNP; hesc.chromHmm.bed表示基于来自ENCODE的ChIP-seq实验的人胚胎干细胞基因组中每个区间的预测功能(通过chromHMM)。
Bedtools的基本overview
Bedtools 是一行命令,只要打bedtools就会出现manual的页面
bedtools
当你输入完bedtools,你会看到有很多subcommands,为了让bedtools成功运行你还需要给它输入一个subcommand,例如:
#这三个是最经典的,下面会着重介绍
bedtools intersect
bedtools merge
bedtools subtract
查看版本
bedtools --version
bedtools "intersect"
Bedtools intersect 是bedtools工具套装中的“主力选手”, 它比较两个或多个BED / BAM / VCF / GFF文件,并识别gemome中两个文件中的特征重叠的所有区域(即共享至少一个碱基对)。
大概的处理方式可以通过下图来理解:
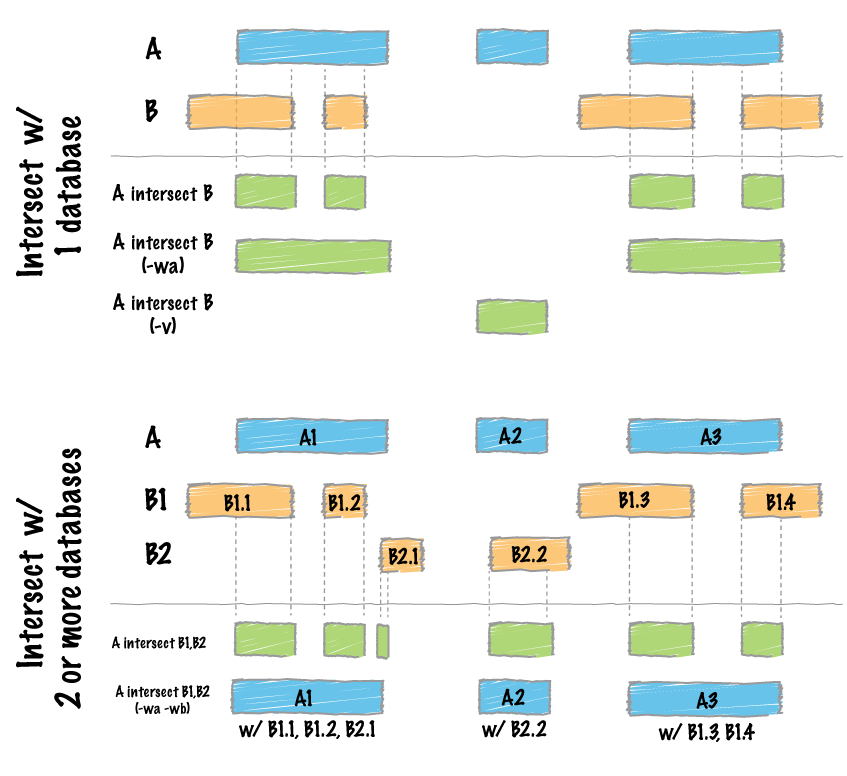
默认的工作方式
默认情况下,intersect报告表示两个文件之间重叠的间隔。为了演示,让我们识别所有与外显子重叠的CpG岛。
bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
在这种情况下,terminal中报告的间隔不是原始的CpG间隔,而是仅仅反映每个原始CpG间隔的与外显子重叠的部分的间隔。
报告每一个文件中其初始的特征
两个 -wa(写入A)和-wb(写入B)选项允许人们查看重叠的A和B文件中的原始记录。因此,它不仅可以显示交叉点的位置,还可以显示相交的位置。
bedtools intersect -a cpg.bed -b exons.bed -wa -wb \
| head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r -
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +
如果你想知道两个bed files中重合的部分,可以使用-wooption:
###在输出文件的最后一行会显示每一个重叠区域的重叠bases的数目。
bedtools intersect -a cpg.bed -b exons.bed -wo \
| head -10
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r - 50
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 439
chr1 713984 714547 CpG:_60 chr1 713663 714068 NR_033908_exon_6_0_chr1_713664_r 0 84
chr1 762416 763445 CpG:_115 chr1 761585 762902 NR_024321_exon_0_0_chr1_761586_r - 486
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_015368_exon_0_0_chr1_762971_f + 185
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047519_exon_0_0_chr1_762971_f + 185
chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047520_exon_0_0_chr1_762971_f + 185
对于“A”文件中的每个特征,我们还可以计算“B”文件中重叠特征的数量。这是使用-c option处理的。
bedtools intersect -a cpg.bed -b exons.bed -c \
| head
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
chr1 533219 534114 CpG:_94 0
chr1 544738 546649 CpG:_171 0
chr1 713984 714547 CpG:_60 1
chr1 762416 763445 CpG:_115 10
chr1 788863 789211 CpG:_28 9
我们通常希望在A文件中识别那些不与B文件中的功能重叠的片段。在这种情况下,-voption是你最好的朋友。
bedtools intersect -a cpg.bed -b exons.bed -v \
| head
chr1 437151 438164 CpG:_84
chr1 449273 450544 CpG:_99
chr1 533219 534114 CpG:_94
chr1 544738 546649 CpG:_171
chr1 801975 802338 CpG:_24
chr1 805198 805628 CpG:_50
chr1 839694 840619 CpG:_83
chr1 844299 845883 CpG:_153
chr1 912869 913153 CpG:_28
chr1 919726 919927 CpG:_15
在默认情况是报告A和B中的片段之间的重叠,只要存在至少一个重叠碱基对即可。但是,-f选项允许您在报告之前指定A中每个偏度的哪个部分应与B中的要素重叠。
假如我们想让整个筛选更加严格,如需要50%的重叠才report出来。
bedtools intersect -a cpg.bed -b exons.bed \
-wo -f 0.50 \
| head
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 439
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047519_exon_5_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047521_exon_4_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047523_exon_3_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047524_exon_3_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047525_exon_4_0_chr1_788771_f 0 348
chr1 788863 789211 CpG:_28 chr1 788858 794826 NR_047520_exon_6_0_chr1_788859_f 0 348
到目前为止,所提出的例子都使用了传统的bedtools算法来寻找交叉点。然而,事实证明,使用预先排序的数据时,bedtools要快得多。
这里的sorted一般是通过sort染色体的名词和其对应序列的顺序,一般染色体信息在第一行,使用sort -k1,1 -k2,2n。
下面就和大家通过一个加单的测试来验证是不是sorted的bed file用bedtools处理起来更加快。
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed > /dev/null
1.10s user 0.11s system 99% cpu 1.206 total
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed -sorted > /dev/null
0.36s user 0.01s system 99% cpu 0.368 total
由于测试文件比较小,所以你可能感觉不出很大差别,但当你处理一个上百G的文件时候,你就会体会到sorted的文件处理速度的提升。
一次intersecting多个文件
在bedtools2.21版本之后,bedtools开始支持一个A文件,一次intersecting多个B文件这样的功能。
这极大地简化了涉及与给定实验相关的多个数据集的分析。例如,让我们将外显子与CpG岛,GWAS SNP和ChromHMM注释相交。
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted | head
chr1 11873 11937 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 11937 12137 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12137 12227 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 +
chr1 13220 14137 NR_046018_exon_2_0_chr1_13221_f 0 +
chr1 14137 14409 NR_046018_exon_2_0_chr1_13221_f 0 +
chr1 14361 14829 NR_024540_exon_0_0_chr1_14362_r 0 -
chr1 14969 15038 NR_024540_exon_1_0_chr1_14970_r 0 -
chr1 15795 15947 NR_024540_exon_2_0_chr1_15796_r 0 -
chr1 16606 16765 NR_024540_exon_3_0_chr1_16607_r 0 -
现在默认情况下,这并不是非常有用,因为我们无法分辨三个“B”文件中的哪一个与每个外显子产生交集。但是,如果我们使用-wa和-wboption,我们可以看到交叉点来自哪个文件编号(遵循命令行上给出的文件的顺序,如下面,1对应cpg.bed,2对应gwas.bed以此类推)。在这种情况下,第7列反映了此文件编号。
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb \
| head -10000 \
| tail -10
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + 3 chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + 3 chr1 27635013 27635413 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27632613 27632813 6_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27632813 27633213 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + 3 chr1 27635013 27635413 7_Weak_Enhancer
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + 1 chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + 3 chr1 27648613 27649413 1_Active_Promoter
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + 1 chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + 3 chr1 27648613 27649413 1_Active_Promoter
当然如果你觉得这样很麻烦,总要看1,2,3是对应哪个文件,你也可以自定义标记你的文件(一般使用文件名缩写)。
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -names cpg gwas chromhmm \
| head -10000 \
| tail -10
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27635013 27635413 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27632613 27632813 6_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27632813 27633213 7_Weak_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27633213 27635013 5_Strong_Enhancer
chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f 0 + chromhmm chr1 27635013 27635413 7_Weak_Enhancer
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + cpg chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f 0 + chromhmm chr1 27648613 27649413 1_Active_Promoter
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + cpg chr1 27648453 27649006 CpG:_63
chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f 0 + chromhmm chr1 27648613 27649413 1_Active_Promoter

如果你有耐心看到这里,我会给你分享一个我最近发现蛮有趣的“智能”文献搜索网页,这个网站允许你上传你的还没写完的paper,或者你阅读的paper,然后这个网站就会给你分析这文章的关键词,最后给你推荐一系列类似的文章。在搜索后,可以自行调整关键词,还可以调整其相关程度,除了关键词外,还支持一系列其他的特征搜索。使用起来体验还不错!
链接如下: https://www.jstor.org/analyze/
大家就慢慢去耍吧,今天分享到此结束。









网友评论