当一个 NGS 项目中对同一个样本进行了 WGS 和 RNA-seq 测序,或者对同一个肿瘤患者的肿瘤组织和癌旁组织同时进行 WGS / WES / RNA-seq / Chip-seq 测序,或者由于第一次测序数据量不够而进行加测得到了两批数据,为了排除实验过程中可能的样本弄混或者标签贴错,就需要验证这不同维度或不同批次的数据是否相互匹配。NGSCheckMate 就是这样一款数据质控软件,可以满足以上所有的需求。
为了判断不同测序数据之间的关系,我之前用过 Mendel(Kinship),KING ,VCFtools (relatedness2 ),但是由于软件接受的输入数据格式问题或者输出结果不直观,都不是很好用,直到我发现了 NGSCheckMate 。NGSCheckMate 可以接受 Fastq/BAM/VCF 作为输入文件,输出文件包含样本聚类关系图,样本之间的相关性系数,样本是否配对说明,比之前我用过的其他软件友好多了。
NGSCheckMate 运行需要 samtools 在系统路径中, 安装之后运行也很简单,以 VCF 输入文件为例:
python ncm.py -V –l INPUT_FILE_list -bed BED_FILE -N test_output –O OUTPUT_DIR
# -l 参数,INPUT_FILE_list 包含所有输入 VCF 文件的绝对路径
# -bed 参数,BED_FILE 软件本身自带,包括使用的所有 SNP marker 位点
# -N 参数,test_output 输出文件前缀
# -O 参数,OUTPUT_DIR 指定输出路径
NGSCheckMate 的文章是 2017 年 3 月份发表在 Nucleic Acids Research 上的,影响因子 10.162。
摘要
在很多利用 NGS 技术的研究中,会对同一个人的不同组织进行测序,或者对同一个组织进行不同水平的测序,例如 DNA-seq,RNA-seq,Chip-seq。在这种项目中需要添加一个质控环节,来确保不同的数据来自同一个人。我们开发了一款简洁易用的软件 NGSCheckMate,利用 SNP 基因型来鉴定数据来源,它可以利用多种格式的数据,如 Fastq,BAM,VCF 来验证不同的数据是否来自同一个人。软件利用已知的单核苷酸多态性位点(SNP)基因型和等位频率信息,即使测序深度不同等位频率不会出现太大波动,以此来鉴定数据是来自同一个体还是分别来自不同的人。我们的测试表明,NGSCheckMate 适用于多种不同水平的数据,包括 WES、WGS、RNA-seq、Chip-seq、靶向捕获测序和单细胞全基因组测序,并且在测序深度低至 0.5X 时依然表现良好。软件有免比对模式,可以直接利用 Fastq 原始数据快速进行数据来源鉴定的模块。我们推荐在 NGS 项目中使用这款工具进行数据质控。软件可以通过 github 项目主页获取。
背景介绍
在很多利用 NGS 技术的研究中,会比较或者整合来源于同一个人的多次测序数据,例如对不同的组织或同一组织不同生理状态下的取样进行测序,比较相同的基因组水平下不同组织间转录组水平的变化,或者检测组织特异性的突变(例如 somatic)。例如在一些癌症研究项目中,同一个患者的肿瘤组织和癌旁组织都会进行 WGS 和 RNA-seq 测序,来检测肿瘤中的体细胞突变及其对基因表达水平的影响。还有一些情况也需要鉴定数据是否来之同一个体,例如重复实验,或者合并不同 lane 上测得的数据(不同 run?)。
质控检查样本与标签是否匹配对于数据分析的重要性不言而喻,特别是对于应用于临床疾病的项目,患者可能需要根据数据分析的结果制定治疗方案。尽管我们有标准的实验质控和数据质控环节,但是在手工操作环节仍有可能出现标签与实际样本不匹配的情况,一旦出现这样的情况,要么丢弃这个可疑样本的数据,要么只能重头开始。样本配对检测应该作为一个数据 QC 环节添加进流程中,而且位于流程的越上游越好。
检测数据是否跟一个个体匹配有多种方法,例如微卫星序列(STR)检测,TCGA 数据库就是利用 PCR 检测 CODIS 数据库中人群高度多态性 STR 的方法来验证肿瘤组织和对应的对照组织是否配对。但是检测 STR 的方法对于 NGS 项目来说并不是很好用,由于大部分 STR 都位于基因组的非编码区,因此对于 RNA-seq 和 WES 测序不适用,另外很多 STR 序列的长度都比 NGS 的测序读长要长。
还有一种比较普遍的方法,就是利用人群高度多态性的 SNP 位点来验证两组数据是否配对。利用 SNP 位点来验证数据已经用于 RNA-seq 和 WES 数据中。NGS 项目中一个人的数据可以来之不同的 lane,还有利用 SNP 位点来检验不同 lane 产出的数据是否来之同一个人的研究。另外,VCFtools 软件也提供了一个功能(relatedness2 模块),可以利用 VCF 文件检验两个个体之间的亲缘系数。但是目前这些方法都只适用于特定的数据类型,不能用来验证不同类型的数据是否来源同一个人。
因此,我们开发了 NGSCheckMate ,一个数据配对 QC 软件,适用于多种数据类型:原始数据 FASTQ、比对数据 BAM 和变异数据 VCF。此外 NGSCheckMate 也支持多组学数据之间的配对检测,例如 WES 数据和 RNA-seq 数据。还有一个免比对功能,可以直接利用两组 FASTQ 数据检测是否匹配。NGSCheckMate 可以利用多种类型的数据,不同测序深度的数据(> 0.5X),而且运行速度非常快,适用范围也很广。
NGSCheckMate 流程和原理
下图展示了 NGSCheckMate 的基本流程, 输入文件支持三种格式:Fastq、BAM、VCF,输出文件也有三个:一个文本文件,包含样本是否配对检测结果和基因型相关性系数;一个 PDF 文件,包含所有样本聚类关系图;一个样本聚类关系 XGMML 文件,可以导入可视化软件中查看,例如 Cytospace。
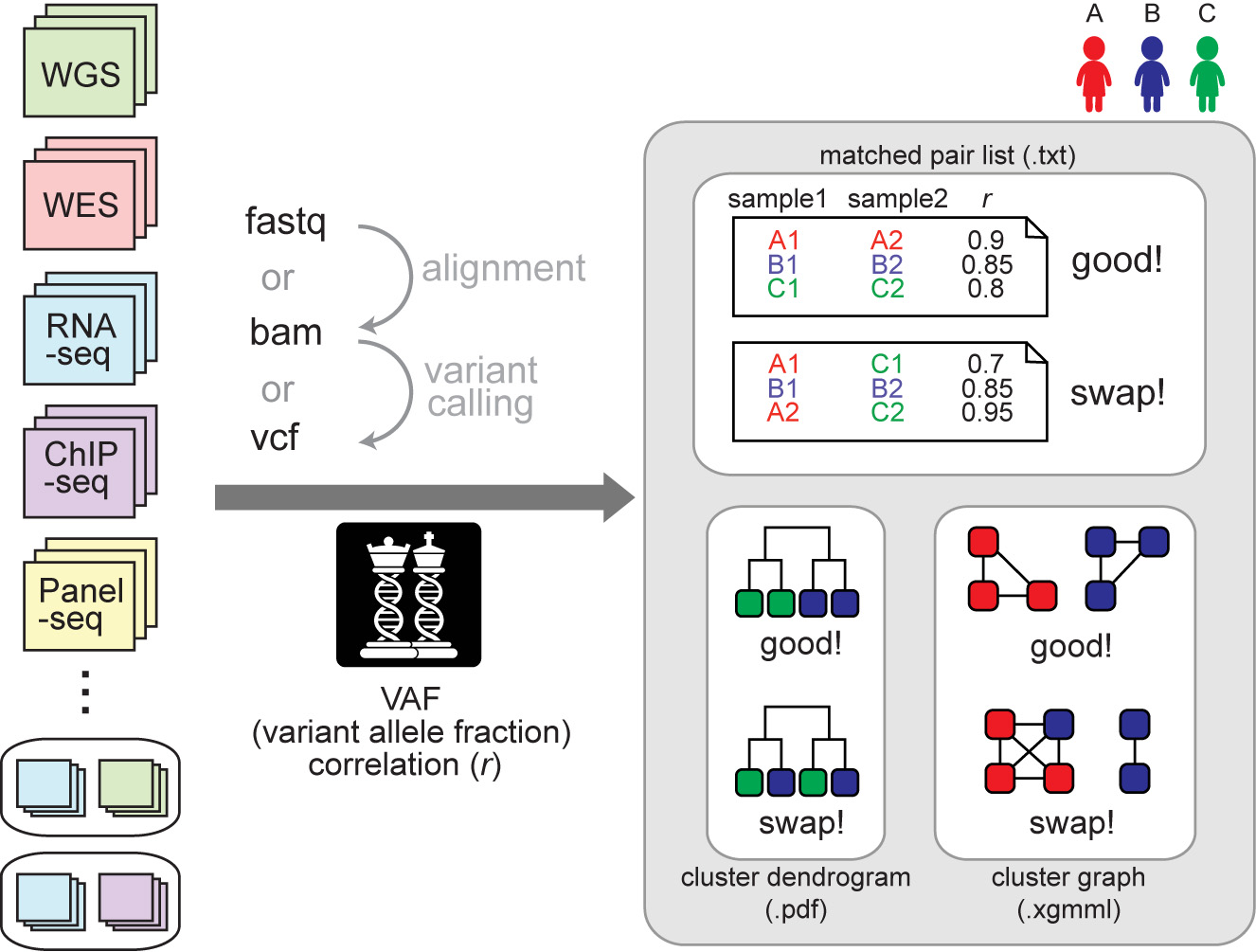 NGSCheckMate 流程图
NGSCheckMate 流程图
NGSCheckMate 通过计算两组输入数据在某些 SNP 位点的 VAF(variant allele fraction) 皮尔森相关系数来判断是否来源同一个人。
VAF = 支持 ALT 的 reads 数量 / 覆盖该 SNP 位点的所有 reads 数量
下图展示了 NGSCheckMate 计算原理。如果输入的是 BAM 文件,NGSCheckMate 会利用 SAMtools mpileup 默认参数来计算 VAF;如果输入的是 FASTQ 文件,软件会自己搜索覆盖 SNP 位点的 reads ,并统计支持 ALT 的 reads 数量和支持 REF 的 reads 数量,以此计算 VAF,当然耗时会长些。
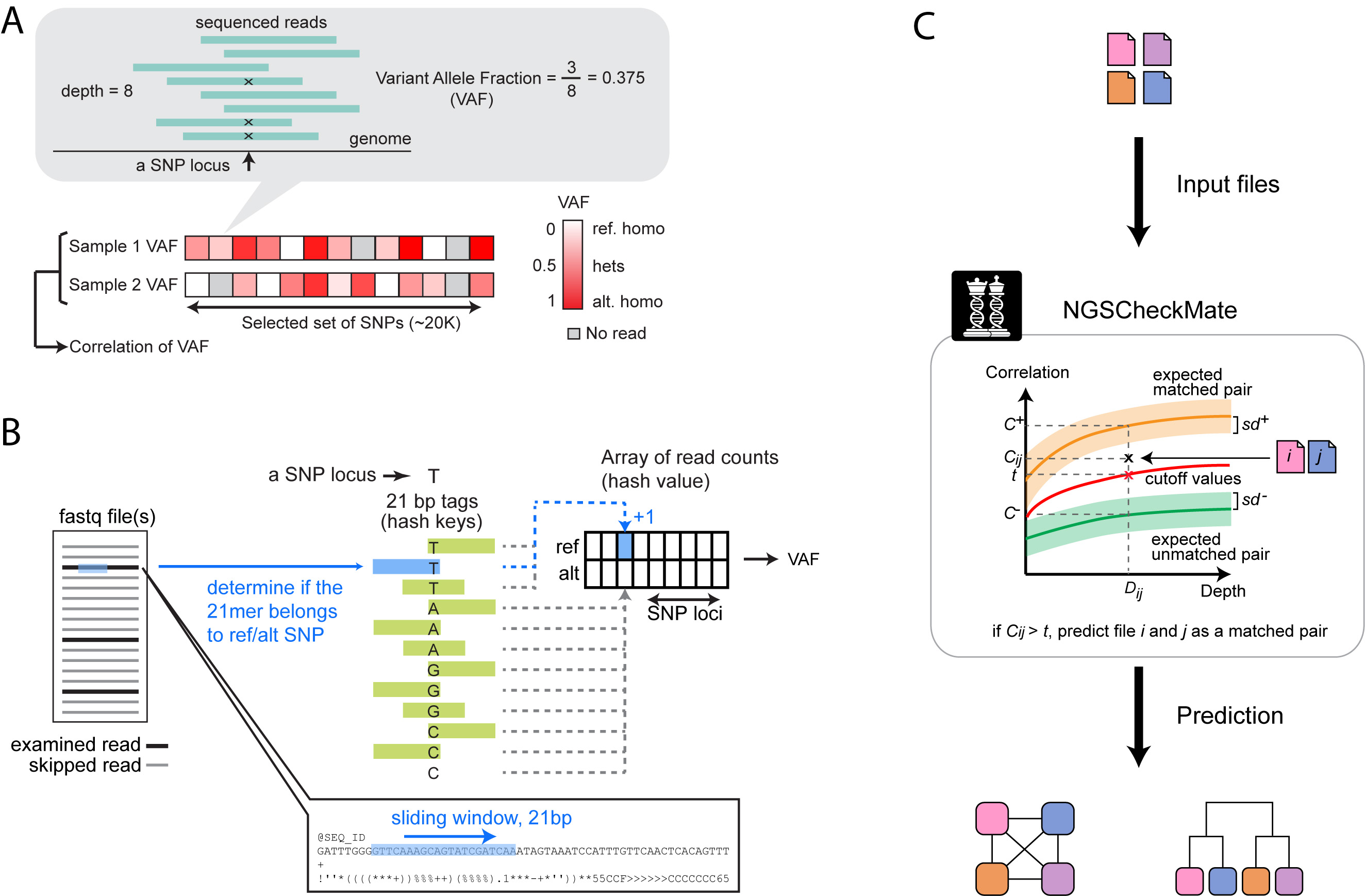 NGSCheckMate 计算原理
NGSCheckMate 计算原理
NGSCheckMate 有一个通过配对和不配对的 WGS 数据训练好的 VAF 相关性系数分布模型,包括 0.01~60X 的测序深度下的相关性系数分布情况,及配对和不配对的相关性系数阈值,只需要将输入数据计算的 VAF 相关性系数与模型分布比较即可得出样本是否匹配的结论。
选择 SNP 作为鉴别不同个体的 marker
利用 TCGA 中 40 对 WGS 数据测试了 dbSNP138 的所有 SNP 位点之后,我们选择了 21067 个外显子区的 SNP 位点作为鉴别不同个体的 marker 集。对于免比对模式(以 FASTQ 为输入),是利用其中 11696 个 SNP 位点作为 marker 集。当然模拟数据显示,只使用其中 11696 个 SNP 位点也可以得到与 21067 个 SNP 相同的 VAF 相关性分布。
免比对模式
免比对模式下,NGSCheckMate 对 reads 取 21bp k-mer 搜索,检测是否能与 SNP 附近的参考序列比对上(包括 ALT 和 REF 状态),为了确保 21bp k-mer 是唯一比对的,免比对模式下把所有参考基因组上下文不唯一的 SNP (SNP 附近的序列在基因组上有多处相同)位点都去掉了。此外为了提高效率,免比对模式会从输入的 FASTQ 中随机下采样,保证足够的准确率同时缩短运算时间。
构建 VAF 相关性系数分布模型
我们在 TCGA 数据库中选取了 40 对的配对样本数据和非配对样本数据,并对它们进行下采样(低至 0.01X),统计不同测序深度下的 VAF 相关性系数分布情况。得到了一个非常稳健的 VAF 相关性系数分布模型(上图 C 所示),我们就用这个分布模型来预测输入数据是否匹配。
对于输入的一组数据,计算它们 VAF 相关系系数,如果落在模型中相同测序深度下的配对系数的分布区间就预测为配对数据,反之亦然。对于不同测序深度数据,判定是否配对的 VAF 相关性系数阈值也不同,测序深度 <1, [1,2), [2,5), [5,10), >=10 阈值分别是 0.38, 0.41, 0.46, 0.55, 0.61 ;但如果样本都来来自同一个家庭(父母关系或兄弟姐妹关系),阈值会更严格一些,分别是 0.50, 0.54, 0.59, 0.69, 0.76 。如果输入的两组数据测序深度不同,则以低测序深度下的判定结果为准。
验证模型的准确性
从 TCGA 数据库和其他一些研究项目中取 160 对(肿瘤/对照) WGS 数据,984 对 WES 数据,170 对 RNA-seq 数据,85 对 panel-seq 数据,130 对单细胞 WGS 数据, 130 组 Chip-seq 数据,来进行测试。另外,除了 Illumina 平台的数据,也测试了 Ion Torrent 平台的两组数据。测试结果显示,在测序深度足够的情况下(>0.5X),NGSCheckMate 预测样本是否配对的准确度接近 100%。
对于有亲缘关系的个体之间的 VAF 相关性系数分布,我们也统计了 10 个家庭的 36 份 WES 数据,如下图 A 中绿色/紫色的点。下图 A 中显示在测序深度大于 0.5X 时,VAF 分布模型可以清晰地将同一个人的数据,有亲缘关系(父母关系和兄弟姐妹关系)的数据,陌生人的数据分开。有亲缘关系的那些数据,父母关系和兄弟姐妹关系的数据分布几乎是重叠的(当然也无法区分到底是什么关系)。
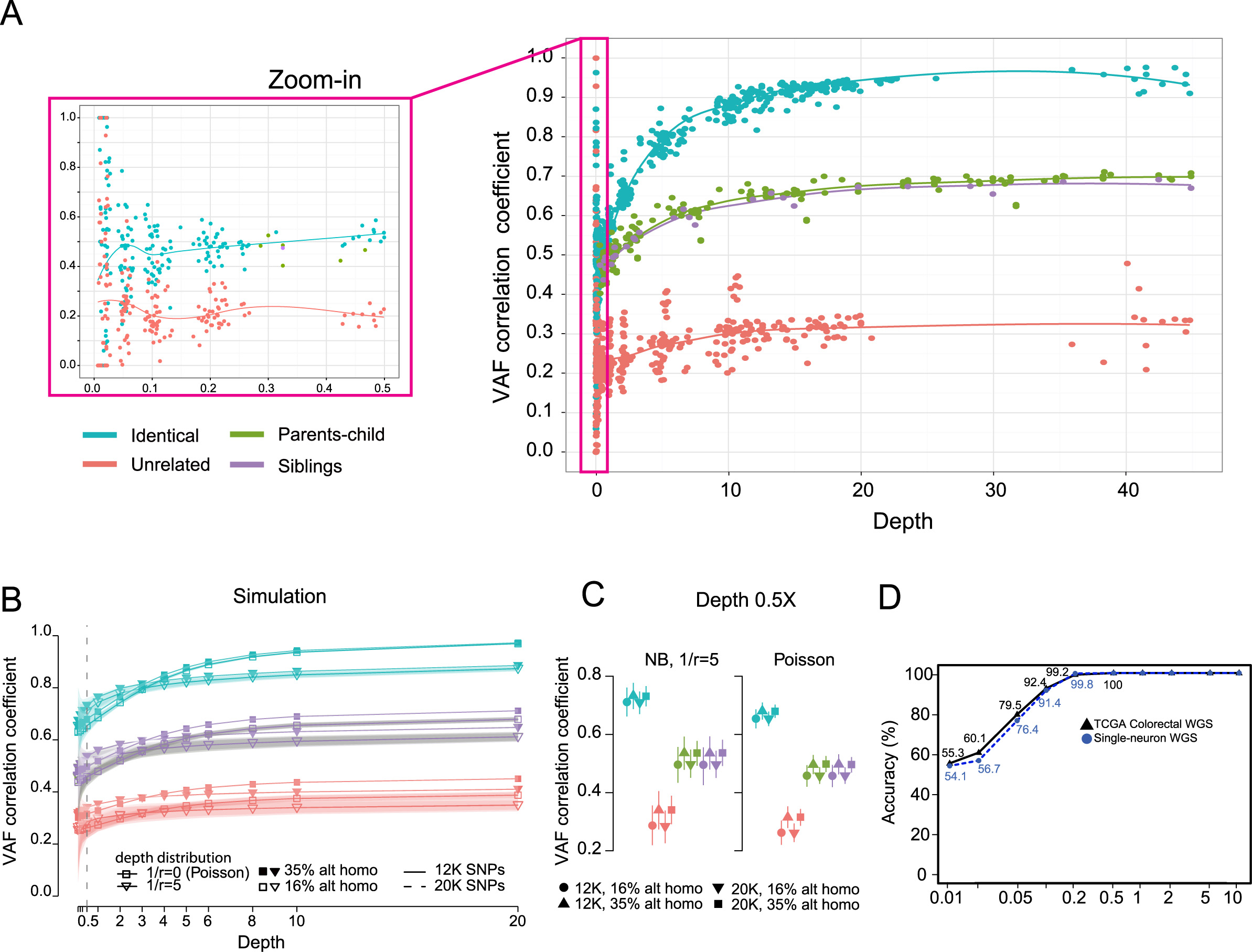 NGSCheckMate-VAF-distributions
NGSCheckMate-VAF-distributions
上图 B 中包含了使用 12K 个 SNP 和使用 20K 个 SNP 计算的 VAF 相关性系数分布,分布非常接近。同时也统计了 SNP 中纯合和杂合的比例对分布模型的影响,分布也几乎是重叠的。
上图 D 中显示了使用 66 对(肿瘤/对照) WGS 数据和 36 对单细胞 WGS 测序数据进行测试的准确度,利用下采样统计不同测序深度下的准确度。在 0.5X 以上测序深度时,模型判断两组数据是否配对的准确度接近 100%。
NGSCheckMate 应用实例:肝癌 WGS 研究项目
在这个项目中对 21 个肝癌患者各自进行 3 种组织(肿瘤,癌旁,血液) WGS 测序,当分析每个患者的体细胞突变 SNV 时候,我们出乎意料地发现有不同的患者 somatic SNV 中有大量是相同的,之后进一步调查,发现可能是部分样本在操作过程中贴错了标签,利用 NGSCheckMate 可以鉴定出所有贴错标签的样本。对于可能弄混的样本重新测序之后,NGSCheckMate 对全部 21 个患者的各自 3 个样本给出了正确的聚类。
此外,利用 NGSCheckMate 也在 TCGA 一项利用低深度 WGS 测序检测胃癌中结构变异的项目中发现 1 例可能是混样的数据(242 个 BAM 文件的其中一个)。
NGSCheckMate 运行速度和内存需求
如果用 VCF 作为输入文件,NGSCheckMate 可以在约 3 分钟之内,利用不超过 200MB 内存,检测 80 个 WGS 测序数据之间的关系。运算速度很快。
如果用 FASTQ 作为输入文件,NGSCheckMate 为了提高速度,会进行下采样(取~2X 数据),也可以在 11 分钟左右,利用约 40 MB内存,检测两个 WGS 原始数据之间的关系。
总结
NGSCheckMate 可以非常高效准确地判定两组数据是否来源与同一个人,适用于多组学数据,包括 WGS,WES,Chip-seq,RNA-seq,panel-seq,而且由于筛选出来的 20K 个 SNP marker 有足够的冗余度,只需要测序深度 >0.5X 就可以给出准确的判断结果。NGSCheckMate 的免比对模式可以在数据下机之后就对 FASTQ 进行检测,尽早发现可能的样本与标签弄混的事故。




网友评论
对肿瘤样本使用多位点 SNP 分型一致性检验,效果更好。
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3978886/