软件介绍
系统发生树是一个很好的工具来推断各种生物之间的进化关系,因此该树已被用于许多进化研究中。因此,基于SNP数据的系统发生树已在重测序项目中成为重点。然而,利用重测序数据确定的大量变异来确定系统发生树并不是一个简单的方法。因此,我们开发了新的管道snphylo,来构建基于snp数据的系统发生树。通过这个管道,用户可以从包含大量SNP数据的文件中构建系统发生树。
特性
- 基于全基因组SNP的树构建。常规树构建基于充满具有某些特性的基因,例如单拷贝基因,核糖体RNA基因,内部转录间隔序列(ITS)。SNPhylo使用全基因组信息构建树,因此,它更准确
- 通过连锁不平衡(LD)减少SNP冗余。同一LD块中的SNP提供冗余谱系信息。SNPhylo在LD块中仅保留一个信息性SNP。它大大减少了运行时间,而不会丢失信息。
- 建树过程高度自动化。 SNPhylo将最常见的SNP /基因型格式(vcf / hapmap)作为输入,并仅使用一个命令生成最大似然树!
流程图
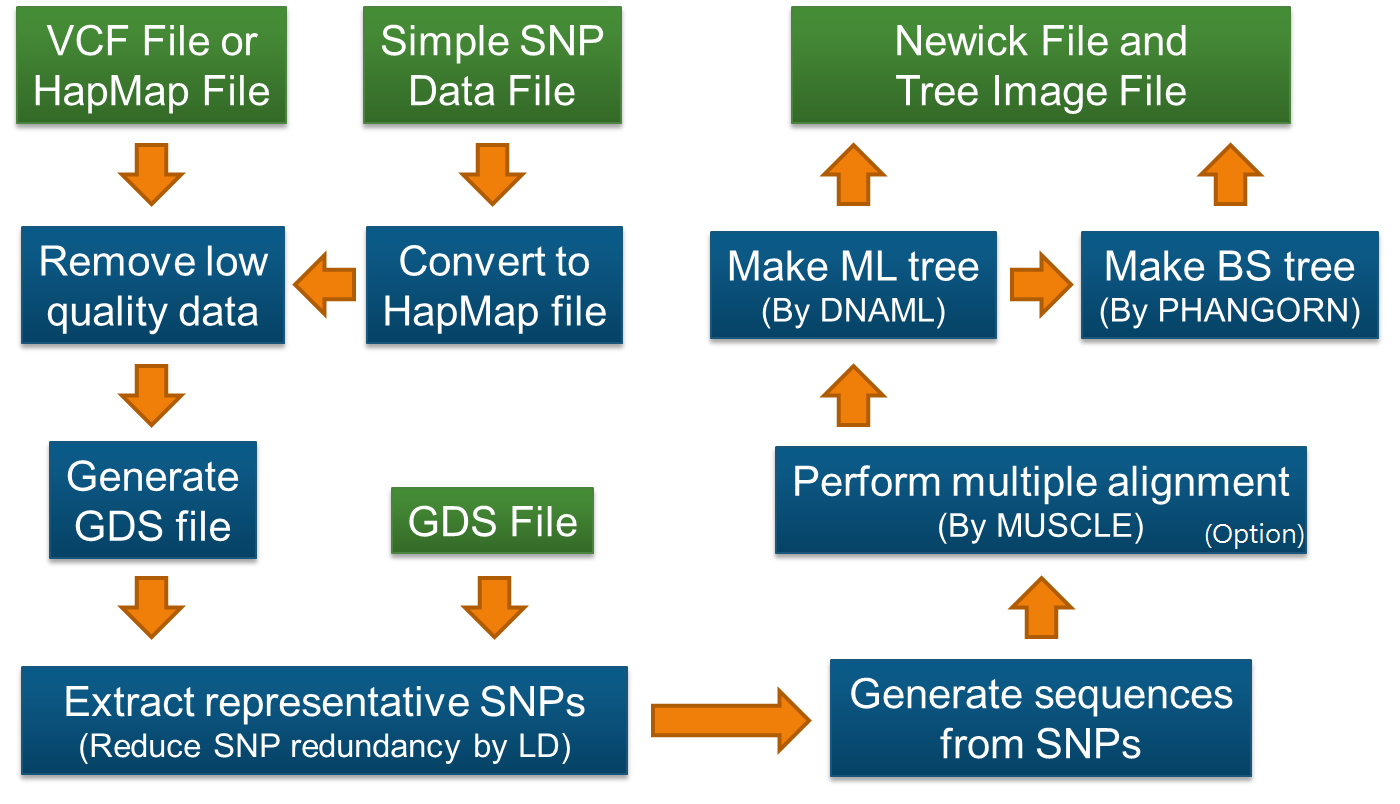 流程图
流程图
下载
安装
相关软件和软件包
Rscript
它包含在软件R中,可从http://www.r-project.org/免费获得。
Python
可以在http://www.python.org/免费获得。
muscle
可以在http://www.drive5.com/muscle/免费获得。
dnaml
它包含在PHYLIP软件中,可从http://evolution.genetics.washington.edu/phylip.html免费获得。
R包(phangorn,gdsfmt,SNPRelate和getopt)
用户可以手动安装这些R包。例如,通过在R中执行以下命令(以root身份),将安装R包。
install.packages(“getopt”,repos =“http://cran.r-project.org”)
install.packages(“phangorn”,repos =“http://cran.r-project.org”)
source(“http://bioconductor.org/biocLite.R”)
biocLite(“gdsfmt”)
biocLite(“SNPRelate”)
安装管道
- 下载文件Snphylo.zip
- 解压缩Snphylo.zip
cd snphylobash setup.sh
设置脚本将找到相关程序,并向您提出一些基本问题来设置SNPlylo。
此外,在用户的许可下,setup.sh可以自动安装在R包之上。
如果设置过程成功完成,您可以看到两个文件(snphylo.sh和snphylo.cfg)。
用法
snphylo.sh -h
snphylo.sh -v VCF_file [-p Maximum_PLCS(5)] [-c Minimum_depth_of_coverage(5)] | -H HapMap_file [-p Maximum_PNSS(5)] | -s Simple_SNP_file [-p Maximum_PNSS(5)] | - d GDS_file [-l LD_threshold(0.1)] [-m MAF_threshold(0.1)] [-M Missing_rate(0.1)] [-o Outgroup_sample_name] [-P Prefix_of_output_files(snphylo.output)] [-b [-B The_number_of_bootstrap_samples(100) )]] [-a The_number_of_the_last_autosome(22)] [-r] [-A] [-h]
选项:
-A:通过MUSCLE执行多重对齐
-b:执行(非参数)引导程序分析并生成树
-h :显示帮助并退出
-r:跳过删除低质量数据的步骤(忽略-p和-c选项)
缩略语:
PLCS:低覆盖率样本的百分比
PNSS:没有SNP信息的样品百分比
LD:连锁不平衡
MAF:轻微等位基因频率
一个基本的执行策略
snphylo.sh -v test.vcf -r -A -b




网友评论